Презентація з біології та екології для учнів 10 класу (рівень стандарту) "Медична генетика"
Презентація з біології та екології для учнів 10 класу (рівень стандарту) "Медична генетика" допоможе роз'яснити наступні питання: медична генетика, основні завдання медичної генетики, спадкові захворювання та хвороби зі спадковою схильністю, медико-генетичне консультування, генотерапія


Медична генетика
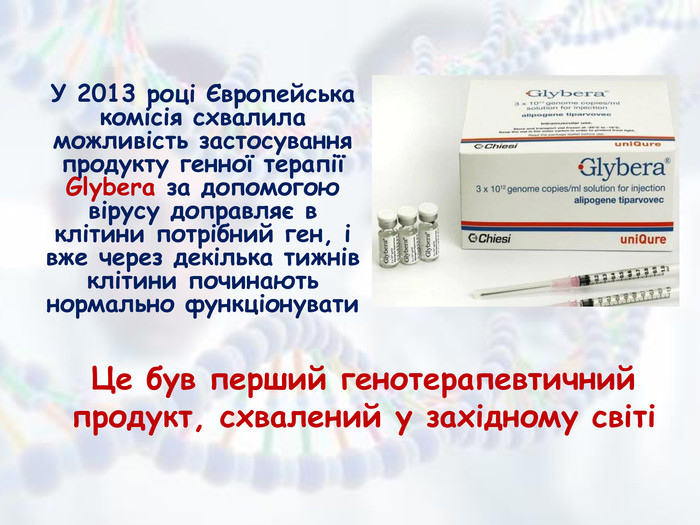
У 2013 році Європейська комісія схвалила можливість застосування продукту генної терапії Glybera за допомогою вірусу доправляє в клітини потрібний ген, і вже через декілька тижнів клітини починають нормально функціонувати. Це був перший генотерапевтичний продукт, схвалений у західному світі
План. Медична генетика. Основні завдання медичної генетики. Спадкові захворювання та хвороби зі спадковою схильністю. Медико-генетичне консультування. Генотерапія

Медична генетика{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}розділ генетики людини, що вивчає роль спадкових чинників у розвитку захворюваньможливості медичної генетики розширені завдяки розвитку молекулярної генетики, біології клітини, молекулярної біології та розуміння біохімічних та молекулярно-генетичних процесів – основи спадкових захворювань;{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Сучасні медико-генетичні дослідження здійснюються на рівнях:молекулярному;клітинному;організмовому;популяційно-видовому{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Методи: полімеразно-ланцюгових реакцій, моделювання

Основні завдання медичної генетики{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}виявлення та опис спадкових захворювань; на сьогодні описано 2200 аутосомно-домінантних, 1420 аутосомно-рецесивних, 286 зчеплених зі статтю генних та близько 8000 хромосомних захворювань;визначення ролі генотипу й чинників середовища в розвитку полігенних (мультифакторіальних) захворювань (ішемічної хвороби, хвороб серця, цукрового діабету, виразкової хвороби, гіпертензії тощо;розшифрування природи великої кількості спадкових захворювань обміну речовин і розроблення ефективних методів профілактики й лікування (наприклад, непереносимість лактози, фенілкетонурія);пізнання складної природи схильності до онкологічних захворювань та механізм реалізації;розроблення дослідницьких програм й методик для розширення можливостей медико-генетичного консультування;профілактика та виліковування спадкових хвороб за допомогою новітніх технологій зміни генів та методів генотерапії

Спадкові захворювання{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}ГенніХромосомніпов’язані із наявністю патологічного алеля певного гена (точкові мутації)пов’язані зі змінами в структурі хромосом (зміна послідовності чи кількості генів у хромосомах) – хромосомні мутаціїможуть успадковуватись за домінантним або рецесивним типомпов’язані з геномними мутаціями - змінами на рівні геному (кількості окремих хромосом або всього набору)зміни ДНК на рівні окремих генівсиндроми котячого лементу, Дауна, Патау, Едвардса – в аутососмахпрогерія, серпуватоклітинна анемія, ферментопатії (фенілкетонурія)пов’язані зі статевими хромосомами: синдроми Клайнфельтера, Тернера, Джейкобса (Якобса)хвороби, пов’язані з дефектами генів, розташованими у Х-хромосомі: дальтонізм, гемофілія
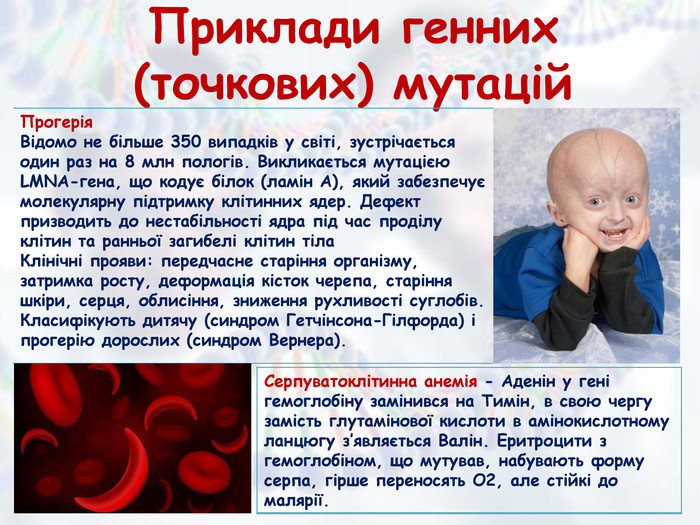
Приклади генних (точкових) мутацій{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Прогерія. Відомо не більше 350 випадків у світі, зустрічається один раз на 8 млн пологів. Викликається мутацією LMNA-гена, що кодує білок (ламін А), який забезпечує молекулярну підтримку клітинних ядер. Дефект призводить до нестабільності ядра під час проділу клітин та ранньої загибелі клітин тіла. Клінічні прояви: передчасне старіння організму, затримка росту, деформація кісток черепа, старіння шкіри, серця, облисіння, зниження рухливості суглобів. Класифікують дитячу (синдром Гетчінсона-Гілфорда) і прогерію дорослих (синдром Вернера).{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Серпуватоклітинна анемія - Аденін у гені гемоглобіну замінився на Тимін, в свою чергу замість глутамінової кислоти в амінокислотному ланцюгу з’являється Валін. Еритроцити з гемоглобіном, що мутував, набувають форму серпа, гірше переносять О2, але стійкі до малярії.
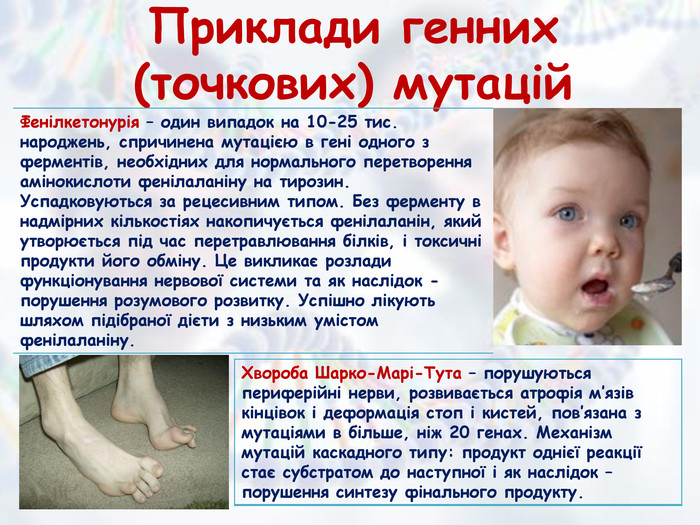
Приклади генних (точкових) мутацій{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Фенілкетонурія – один випадок на 10-25 тис. народжень, спричинена мутацією в гені одного з ферментів, необхідних для нормального перетворення амінокислоти фенілаланіну на тирозин. Успадковуються за рецесивним типом. Без ферменту в надмірних кількостіях накопичується фенілаланін, який утворюється під час перетравлювання білків, і токсичні продукти його обміну. Це викликає розлади функціонування нервової системи та як наслідок - порушення розумового розвитку. Успішно лікують шляхом підібраної дієти з низьким умістом фенілаланіну.{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Хвороба Шарко-Марі-Тута – порушуються периферійні нерви, розвивається атрофія м’язів кінцівок і деформація стоп і кистей, пов’язана з мутаціями в більше, ніж 20 генах. Механізм мутацій каскадного типу: продукт однієї реакції стає субстратом до наступної і як наслідок – порушення синтезу фінального продукту.

Приклади хромосомних мутацій{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Синдром котячого лементу. Зустрічається в 1 з 50000 новонароджених. Розвивається в результаті делеції кінцевої ділянки п’ятої хромосоми. Клінічні прояви: порушення функцій серцево-судинної, травної систем, недорозвинення гортані (з характерним криком, що нагадує котяче нявкання), загальне відставання розвитку, розумова відсталість, місяцеподібне обличчя з широко розставленими очима.{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Нейсприйнятливість до ВІЛЗустрічається у 10% європейців. Пов’язана ізделецією в гені, що кодує рецептор ССR5, який використовується вірусом імунодефіциту людини (ВІЛ) для розпізнавання Т-лімфоцитів. Продукт гену з делецією отримав назву CCR5-Δ32, та не розпізнається ВІЛ, і носії такої мутації до ВІЛ несприйнятливі.

Приклади геномних мутацій{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Синдром Дауна 47, 21+ (трисомія 21 пари хромосом)1 випадок на 700 - 1000 пологів;ризик народження зростає з віком матері;Клінічні прояви:вкорочений череп, розкосі очі, маленький ніс та вуха, характерна складка очей епікамп, аномалії зубів, збільшений язик;вроджені вади серця та травної системи;м’язова гіпотонія;дисфункція щитоподібної залози;порушення зору й слуху;маленький зріст, короткі кінцівки та вкорочені пальці;розумова відсталість;при належному догляді доживають до 60-70 років{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Наслідками геномних мутацій можуть бути синдроми або смерть

Приклади геномних мутацій{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Синдром Патау 47, 13+ (трисомія 13 пари хромосом)1 випадок на 12000-29000 вагітностей. Клінічні прояви:низька вага,деформація черепа, дефекти щелеп, мозок не ділиться на 2 півкулі;аплазія (відсутність шкіри на ділянках голови);полідактилія;вади розвитку статевих органів;відхилення у фізичному та розумовому розвитку;1% дітей доживають до 10 років. Синдром Едвардса 47, 18+ (трисомія 18 пари хромосом)1 випадок на 5000-7000 вагітностей. Клінічні прояви:низька вага;деформація черепа, дефекти щелеп;вади розвитку серцево-судинної, сечовидільної, травної систем, опорно-рухової системи;відсутність фізичного та розумового розвитку;90% дітей не доживають до 1 року
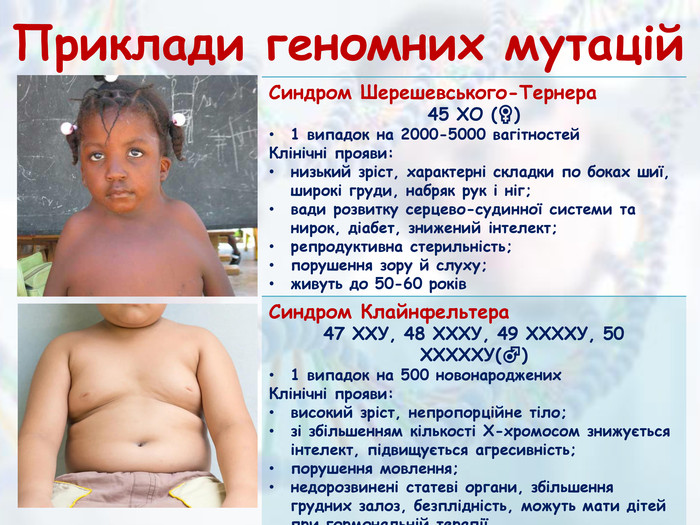
Приклади геномних мутацій{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Синдром Шерешевського-Тернера 45 ХО (♀)1 випадок на 2000-5000 вагітностей. Клінічні прояви:низький зріст, характерні складки по боках шиї, широкі груди, набряк рук і ніг;вади розвитку серцево-судинної системи та нирок, діабет, знижений інтелект;репродуктивна стерильність;порушення зору й слуху;живуть до 50-60 років. Синдром Клайнфельтера 47 ХХУ, 48 ХХХУ, 49 ХХХХУ, 50 ХХХХХУ(♂)1 випадок на 500 новонароджених. Клінічні прояви:високий зріст, непропорційне тіло;зі збільшенням кількості Х-хромосом знижується інтелект, підвищується агресивність;порушення мовлення;недорозвинені статеві органи, збільшення грудних залоз, безплідність, можуть мати дітей при гормональній терапії

Приклади геномних мутацій{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Синдром Джейкобс (Якобса)47 ХУУ (♂)1 випадок на 1000 новонароджених хлопчиків. Клінічні прояви:високий зріст, порушення координації рухів;порушення мови, проблеми з навчанням, гіперактивність, поведінкові проблеми (імпульсивні, емоційно нестабільні);фертильність не порушена;обмежень для життя немає{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Лікування хромосомних хвороб є неможливим. Медицина може лише пом’якшити негативний вияв симптомів{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Хвороби із спадковою схильністю{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}успадковується не хвороба, а схильність захворіти (шизофренія, маніакально-депресивний синдром)хвороба може викликатись несприятливими факторами (інфекційні захворювання, шкідливі звички

Лабораторні тварини{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}у вирішенні проблем медичної генетики допомогу надають лабораторні тваринистворено тисячі ліній «нокаутних мишей», які слугують модельними організмами для вивчення хвороб людини;«нокаутна миша» – мутантна миша, в якої вимкнено певні гени;{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}після розшифрування геному Жаби шпоркової (Xenopus tropicalis) вчені виявили близько 80% генів, подібних до тих, що відповідають за спадкові захворювання{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}інактивуючи специфічний ген Миші хатньої (Mus musculus) та досліджуючи зміни фенотипу, можна зробити висновки про роль даного гена;{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}сучасна генетика інтенсивно розширює дослідження в різних напрямах: вивчення геному людини, мітохондріальна генетика, імуногенетика,генетика розвитку, клінічна генетика тощо

Медико-генетичне консультування{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}процес, пов’язаний з вирішенням проблем, що призводять до появи або ризику появи спадкових захворювань у родині;допомога й найбільш поширена форма профілактики спадкових захворювань;{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}дає змогу лікарям діагностувати хворобу та надавати конкретні рекомендації щодо лікування{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Основні завдання медико-генетичних консультаційвстановлення діагнозу спадкового захворювання;пояснення змісту прогнозу;визначення типу успадкування захворювання;спостереження і виявлення груп підвищеного ризику серед родичів;розрахунок ступеню ризику повторення захворювання;поширення медико-генетичних знань серед населення

Медико-генетичне консультування{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Розрахунок ступеню ризику повторення захворюваннягенетичний ризик, що не перевищує 5%, розцінюється як низькийдо 20% - як підвищений;понад 20% - як високий ризик{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Етапи медико-генетичної консультаціїдіагноз;висновок;прогноз;порада{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Необхідним є відверте і доброзичливе спілкування лікаря-генетика з родиною хворого

Генотерапія{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}лікування захворювань шляхом заміни дефектних генів нормальними;основою генотерапії є сукупність методів лікування спадкових, онкологічних, деяких вірусних захворювань шляхом внесення змін у генетичний апарат клітин пацієнтів з метою спрямованої зміни генних дефектів або надання клітинам нових функцій;{BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}першим успіхом генотерапії став випадок виліковування дитини від тяжкого імунодефіциту за ферментом аденозиндезамінази;14 вересня 1990 р. дівчинці було пересаджено власні лімфоцити із попередньо заміненим геномом;Білок аденозиндезамінази{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}згодом були здійснено чергові трансфузії генетично змінених Т-лімфоцитів і загальним результатом стало поліпшення стану пацієнтки та її нормальний спосіб життя
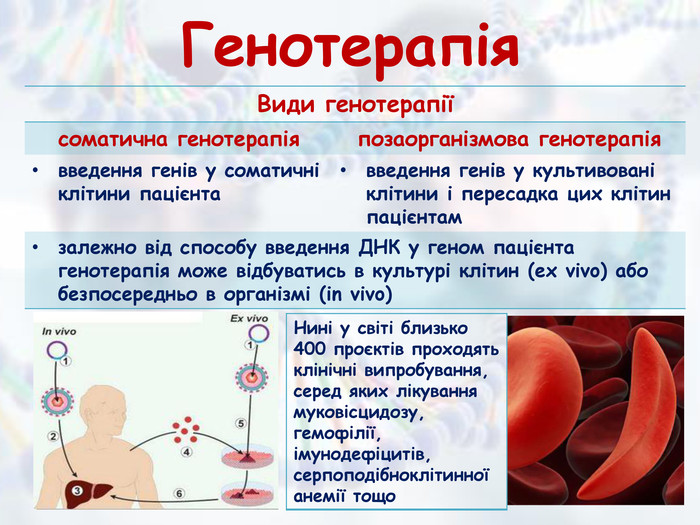
Генотерапія{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Види генотерапіїсоматична генотерапіяпозаорганізмова генотерапіявведення генів у соматичні клітини пацієнтавведення генів у культивовані клітини і пересадка цих клітин пацієнтамзалежно від способу введення ДНК у геном пацієнта генотерапія може відбуватись в культурі клітин (ex vivo) або безпосередньо в організмі (in vivo){BDBED569-4797-4 DF1-A0 F4-6 AAB3 CD982 D8}Нині у світі близько 400 проєктів проходять клінічні випробування, серед яких лікування муковісцидозу, гемофілії, імунодефіцитів, серпоподібноклітинної анемії тощо
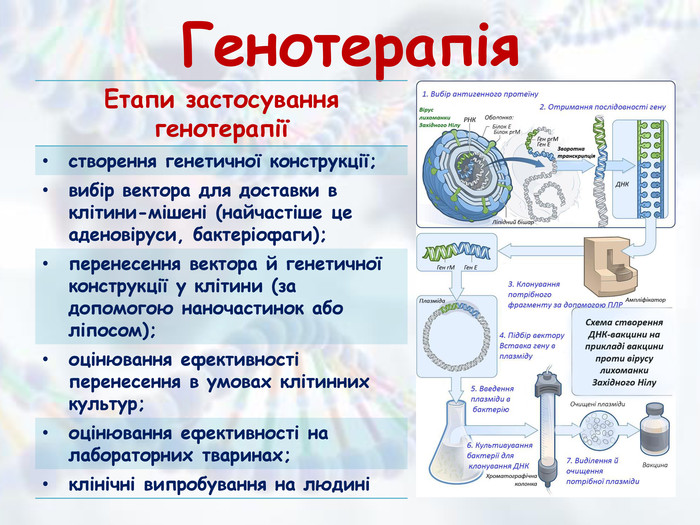
Генотерапія{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}Етапи застосування генотерапіїстворення генетичної конструкції;вибір вектора для доставки в клітини-мішені (найчастіше це аденовіруси, бактеріофаги);перенесення вектора й генетичної конструкції у клітини (за допомогою наночастинок або ліпосом);оцінювання ефективності перенесення в умовах клітинних культур;оцінювання ефективності на лабораторних тваринах;клінічні випробування на людині
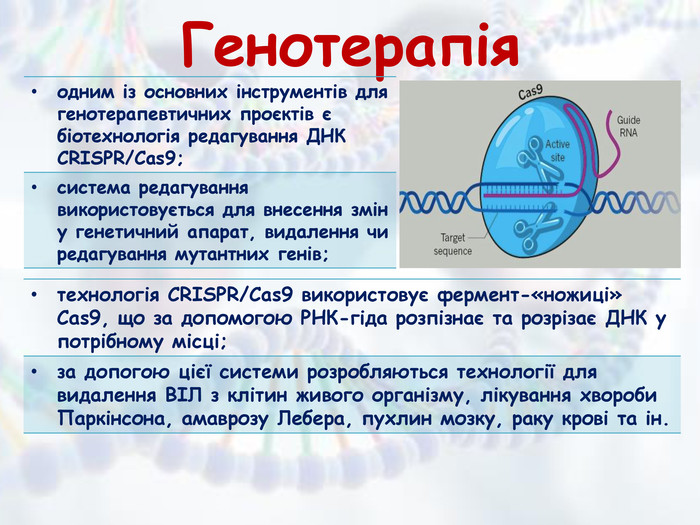
Генотерапія{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}одним із основних інструментів для генотерапевтичних проєктів є біотехнологія редагування ДНК CRISPR/Cas9;система редагування використовується для внесення змін у генетичний апарат, видалення чи редагування мутантних генів;{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}технологія CRISPR/Cas9 використовує фермент-«ножиці» Cas9, що за допомогою РНК-гіда розпізнає та розрізає ДНК у потрібному місці;за допогою цієї системи розробляються технології для видалення ВІЛ з клітин живого організму, лікування хвороби Паркінсона, амаврозу Лебера, пухлин мозку, раку крові та ін.

Генетичні маркери{5 FD0 F851-EC5 A-4 D38-B0 AD-8093 EC10 F338}асоційовані із захворюваннями варіанти генів або інших ділянок ДНК;на сьогодні відомо понад 500 мутантних генів;деякі мутації генів BRCA1 та BRCA2 збільшують ризик розвитку у жінки раку молочної залози та яєчників;виявлені гени, які збільшують ризик виникнення цукрового діабетує варіанти генів, які підвищують ризики розвитку хвороб Альцгеймера, Паркінсонау разі виявлення таких маркерів пропонуються спеціальні програми профілактичних заходів (відповідна дієта та поведінка) з метою істотного покращення життя та продовження його тривалостіХвороба Альцгеймера. Хвороба Паркінсона. Рак яєчників

Висновки. Предметом медичної генетики є спадкові генні й хромосомні хвороби, вади розвитку та хвороби зі спадковою схильністю. Організація медико-генетичного консультування пов’язане з інформуванням людини про ризик розвитку спадкового захворювання, передачі його нащадкам та про діагностичні та терапевтичні діїГенотерапія стає одним із найважливіших інструментів медицини ХХІ століття

Домашнє завдання. Опрацювати §43;с. 167-172 (розглядати рисунки, відповідати на питання усно);Переглядати презентацію на сайті "Дистанційне навчання"

Дякую за увагу!


про публікацію авторської розробки
Додати розробку
-

Бардукова Ніна
22.02.2026 в 13:15
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
-

Машкіна Тетяна Володимирівна
25.02.2024 в 18:18
Дякую за чудову презентацію!!!!
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
-

Колобова Аліна Миколаївна
02.02.2023 в 23:48
Дякую!
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
-

Бабарика Костянтин Віталійович
04.04.2022 в 21:19
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
Показати ще 1 відгук