Спадкові захворювання людини

Тема: Спадкові захворювання людини.
Мета:
навчальна: сприяти формуванню знання про найпоширеніші спадкові захворювання людей; визначити особливості успадкування та вірогідність прояву захворювання в нащадків;
розвивальна: розвивати образне та логічне мислення, удосконалювати навички самостійного навчання; розширити межі наукового світогляду школярів;
виховна: сприяти вихованню доброзичливого, ввічливого, тактовного учня;формувати любов до біології як науки, та відповідальне ставлення до до власного здоров'я та здоров'я майбутніх дітей.
Тип уроку: комбінований
Обладнання: підручник, роздаткові картки, слайди презентації із зображенням прикладів вад розвитку людини,телевізор,комп’ютер.
Структура уроку:
- Організаційний момент – 3 хв.
- Актуалізація опорних знань – 5 хв
- Мотивація навчальної діяльності. Повідомлення теми та мети уроку – 5 хв
- Вивчення нового матеріалу – 20 хв
- Закріплення вивченого матеріалу – 10 хв
- Домашнє завдання – 2 хв
Хід уроку
I. Організаційний момент:
- Доброго дня! Доброго, теплого, дня! Олександр Дюма, видатний французький письменник, коли чекав гостей на звану вечірку, у гардеробі вішав табличку з написом: «Отут, разом з одягом, залишайте свій поганий настрій». Мені б дуже хотілося, щоб і ви залишили негатив, якщо такий є, за дверима класу і налаштувалися на роботу.
На сьогоднішньому уроці ваша оцінка буде формуватися наступним чином:я роздаю вам картки само оцінювання,у вас є три кретерія. 1.Перевірка домашнього завдання,яке оцінюється в 2 бали,оцінка за випереджувальне завдання 5 балів,та вирішування задач для закріплення навичок 5 балів. Ваше завдання підписати ці картки та виставити оцінки,наприкінці уроку ви здасте мені їх,та я виставлю вам оцінки в журнал. Тоді почнемо з 1 етапу,тобто перевірки домашнього завдання.
II. Актуалізація опорних знань
У нас з вами є вже відома нам вправа «Лото»,ваше завдання вибрати клітинку з номером,де зашифроване питання та дати на нього відповідь.Починаємо.
Вправа «Лото»
Мутації,спадковість,мінливість,ген,хромосома,комбінативна мінливість, мутаційна мінливість, норма реакції, генотип, фенотип,локус,домінантна ознака.
Чудово,ми з вами повторили основні терміни,які нам знадобляться сьогодні на уроці. Але перш,ніж оголосити тему,я хочу,щоб ви самостійно дізналися,про що буде йти мова сьогодні на уроці.
III. Мотивація навчальної діяльності. Оголошення теми та мети уроку.
Перед Вами – чорна скринька. Ваше завдання- відгадати, що в ній знаходиться. При цьому необхідно проявити кмітливість, винахідливість і здатність мислити нестандартно. Знавці дають Вам підказки, за якими Ви зможете визначити цей унікальний предмет.( Відкриваю конверт з підказками, зачитую їх):
1.Основу становить дволанцюгова молекула ДНК з ядерними білками-гістонами.
2. Кожен організм має свій видовий набір, який забезпечує передачу спадковості нащадкам.
3. Найкраще її будову видно тоді, коли перебуває в метафазі мітозу. То що ж знаходиться в чорній скриньці? Так, це хромосома. Але в хромосомах можуть відбуватись зміни, які є причиною важких, невиліковних хвороб людини,тож тема нашого уроку Спадкові захворювання людини.
Мета уроку:
- Дати визначення,що таке спадкові захворювання.
- Яка є класифікація спадкових захворювання.
- Ознайомитися з хворобами,що передаються нащадкам.
- З’ясувати,який розділ біології допомагає прогнозувати ці захворювання.
- Розв’язування генетичних задач.
Епіграфом нашого уроку сьогодні будуть наступні слова невідомого автора: Епіграф уроку
Говорячи про генетику. Непогано було б уточнити,
що краще успадкувати таланти, ніж хвороби…
Проте мені здається,що вони найкраще підходять до нашого з вами уроку.
Відкриваємо зошити,записуємо дату та тему уроку.
III. Вивчення нового матеріалу.
Уявімо собі ситуацію:Пролунав весільний марш Мендельсона. Народилася молода сім'я. І ось в один прекрасний момент молодята вирішили завести дитину. Вони мріють про те, що їхня дитина буде красивим, як мама. Розумним, як тато. Папа вимагає сина. А мама хоче дочку. Спори можуть перерости у сварку. І тоді втручається мудра бабуся, яка пояснює, що головне, щоб дитина була здоровою. А й справді, не завжди народження дитини приносить в будинок тільки радість. Чому?
Сумна статистика. У 1986 році було відомо 2 тис. спадкових захворювань, а в 1992 році їх кількість зросла до 5 тисяч. Щорічно в Україні народжується 200 тисяч дітей зі спадковими хворобами. З них 40 тисяч залишаються жити з вродженими вадами. Щороку в світі народжується 90 тисяч розумово відсталих дітей і 150 тисяч тих, кому буде важко вчитися. Чим можна пояснити ці дані? В чому причина?
Усі ми мріємо про те, щоб наші діти були здоровими, здібними та щасливими, щоб від нас, батьків вони отримали лише хороші якості. Але так буває не завжди. І ви вже знаєте, що передаються нащадкам схильність (підвищена ймовірність) захворіти на атеросклероз, ішемічні хвороби серця, діабет, хвороби шлунку, алкоголізм, онкологічних захворювань, а не лише спадкові хвороби. Та й що ми знаємо про спадкові хвороби? Можливо вони з’явилися, як наслідок науково – технічного прогресу?
Проте палеонтологічні знахідки свідчать про те, що такі захворювання існували завжди.
Що ми знаємо взагалі про спадкові захворювання? Давайте дамо визначення. Записуємо в зошит: Спадкові захворювання – це захворювання,зумовлені порушеннями в процесах збереження ,передачі та реалізації генетичної інформації. Відповідно до цього є класифікація спадкових хвороб:
1) Моногенні , або молекулярні. Генетичне порушення пов’язане з мутацією в одиничному локусі хромосоми. Напр. глухота,сліпота.
2) Хромосомні. Пов’язані зі змінною структури хромосом(синдром котячого крику) і кількості хромосом – хвороба Дауна.
3) Полігенні,або мультифакторіальні.
Відповідно до цього ви були розподілені на групи та мали випереджувальне домашнє завдання,щодо характеристики цих захворювань. Тож запрошуємо 1 групу до виступу. Для виступу кожна група має 5 хвилин.
Знайомство зі спадковими хворобами . Виступи учнів, презентації робіт по заданим темам.
Дякую,всі чудово справилися зі своєю роботою. Як ви гадаєте,а чи існує розділ біології ,який допомагає людям вирішити проблеми спадкових захворювань?Так,це розділ біології,називається медична генетика.
Фізкультхвилинка
Медико-генетичне консультування.
В наш час відомо сотні захворювань, у яких механізми біохімічних процесів вивчено досить докладно. Оскільки причиною спадкових хвороб є порушення в структурі генетичного матеріалу, повне лікування їх поки що неможливе. Тому потрібно якомога раніше діагностувати патологію й почати профілактичні та лікувальні заходи.
Спадкові захворювання людини легше попередити, ніж вилікувати. Для профілактики спадкових захворювань створені спеціалізовані медичні центри, які здійснюють генетичне консультування.
Медико–генетичне консультування – це спеціальний вид медичної допомоги населенню, направлений на профілактику спадкових хвороб. В ідеальній ситуації за такою консультацією повинні звернутися всі майбутні батьки, які хочуть мати дитину.(записуємо визначення)
Медико–генетичні консультації допомагають людям у прогнозуванні шлюбу, потомства,консультують вагітних жінок, допомагають у вирішенні питань спадковості, усиновлення дитини, проводять точну діагностику спадкових захворювань. Під час консультування визначається прогноз народження дитини зі спадковою патологією,пояснюється вірогідність цієї події і надається допомога у прийнятті рішення про народження.
Складається з 3-х етапів:
- Уточнення діагнозу із застосуванням спеціальних методів: генеалогічне обстеження й складання родоводу,пренатальна діагностика( УЗД, біохімічний скринінг білків у сироватці вагітної жінки, амніоцентез – аналіз навколоплідної рідини для визначення каріотипу плоду).
- Складання прогнозу для дитини, який базується на даних про тип і варіант успадкування патологічного стану.
- Формування висновку та пояснення генетичного ризику в доступній формі.
Висновок: таким чином ми можемо зробити висновок, що основною умовою народження здорових дітей є міцне здоров’я батьків і відсутність у них генетичних відхилень, та шкідливих звичок.
IV. Закріплення вивченого матеріалу:
Чим більше людина пізнає себе, тим більше виникає питань. Не випадково у генетиків побутує приказка « рух від помилкового знання до дійсного незнання – це вже великий прогрес». Тож давайте й ми зробимо кілька кроків на цьому шляху. До нас надійшли листи від родин з проханням про допомогу. Ми з вами спробуємо побути в ролі генетиків і вирішити їх проблеми. Я вам роздам листи,ваше завдання уважно їх прочитати,та допомогти вирішити проблему. Після 2 – х хвилин часу,капітани команд повинні дати відповідь.Робота в групах.
Задача 1. Ахондроплазія – розповсюджена форма карликовості (хвороба Парро-Марі), успадковується як домінантна ознака. Чоловік з короткими кінцівками одружується з жінкою, що має нормальну будову кінцівок. Яка вірогідність прояву патології у нащадків, якщо його батько мав нормальні кінцівки?
Дано:
А – ген ахондроплазії Р: аа х Аа
а – ген нормальної довжини G: а А
АА – ахондроплазія а
Аа – ахондроплазія F: Аа аа
аа – нормальна будова кінцівок ахо норма
___________________________ 50% нащадків страждатимуть вадою
F - ?
Задача 2. Дефект гіпоплазії (витончення емалі) успадковується по домінантному типу, що зчеплений з Х-хромосомою. Жінка, яка має гіпоплазію , вийшла заміж за чоловіка, який мав такий самий дефект. Від шлюбу народжується здоровий хлопчик. Чи є вірогідність появи здорової доньки в цій сім’ї?
Дано:
ХА – ген гіпоплазії Р: ХАХа х ХА Y
Ха – ген норми G: ХА ХА
ХАХА – жінка з гіпоплазією Ха Y
ХАХа – жінка з гіпоплазією F: ХАХА Х А Х а ХАY Ха Y
ХаХа – жінка норма емалі гіпо гіпо гіпо норма
ХАY – чоловік з гіпоплазією
Ха Y – чоловік з норма емаллю 25% здорових хлопчиків ___________________________ 0% здорових дівчат
F - ? Р - ?
Задача 3. Гемофілія – не згортання крові рецесивна ознака, що пов’язана з Х – хромосомою. Жінка, батько якою страждав гемофілією, одружена зі здоровим чоловіком, звернулись до МГК з проханням визначити вірогідність народження статі здорової дитини? Яку консультацію надав лікар?
Дано:
ХА – ген норми Р: ХАХа х ХА Y
Ха – ген гемофілії G: ХА ХА
ХАХА – жінка норми Ха Y
ХАХа – жінка норми, носій F: ХАХА ХА Х а ХАY Ха Y
ХаХа – жінка гемофілік, летальність норма норма, носій норма гемофілік
ХАY – чоловік норма
Ха Y – чоловік гемофілік 25% здорових хлопчиків
__________________________ 25% здорових дівчаток
F - ? Р - ?
Дяку,ви чудово справилися зі своїм завданнями та допомогли родинам ,які звернулися до нас за допомогою.
V. Підсумок уроку
Рефлексія
Вчитель:
Підсумовуючи наш з вами урок,можна повернутися до нашого епіграфу,та підтвердити,що звичайно краще успадковувати від батьків талант та розумові здібності,але нажаль не завжди виходить саме так . Проте люди,які мають спадкові захворювання,та живуть серед нас,так само заслуговують на повагу та розуміння з нашого боку.
Ми з вами сьогодні плідно порацювали,і наприкінці уроку я хотіла ,щоб ви оцінили нашу роботу. У нас на дошці є прикріплена картинка популярного додатку,яким ви всі користуєтеся. Давайте уявимо,що наш сьогоднішній урок був,як перегляд відео в цьому додатку,щоб ви зробили. Поширили його,поставили відмітку подобається,чи можливо у вас залишилися питання і ви б його прокоментували? Прошу вас та наших гостей оцінити його будь – якою дією.
VІ. Повідомлення домашнього завдання.
Параграф 71. Підготувати по 5 тестових питань для однокласників по сьогоднішній темі. Дякую за увагу,гарного дня!
Додаток до уроку:
Генні патології (молекулярні)
До генних хвороб у людини відносяться численні хвороби обміну речовин. Вони можуть бути пов'язані з порушенням обміну вуглеводів, ліпідів, стероїдів, пуринів і піримідинів, білірубіну, металів та ін Поки ще немає єдиної класифікації спадкових хвороб обміну речовин. Науковою групою ВООЗ запропонована наступна класифікація:
1) хвороби амінокислотного обміну (фенілкетонурія, алкаптонурія та ін.);
2) спадкові порушення обміну вуглеводів (галагоземія, глікогенових хвороба тощо);
3) хвороби, пов'язані з порушенням ліпідного обміну (хвороба Німана-Піка, хвороба Гоше та ін.);
4) спадкові порушення обміну стероїдів;
5) спадкові хвороби пуринового і піримідинового обміну (подагра, синдром Леша-Найя та ін.);
6) хвороби порушення обміну сполучної тканини (хвороба Марфана, мукополісахариди та ін.);
7) спадкові порушення гема-і порфірину (гемоглобінопатії та ін..);
8) хвороби, пов'язані з порушенням обміну в еритроцитах (гемолітичні анемії та ін..);
9) спадкові порушення обміну білірубіну;
10) спадкові хвороби обміну металів (хвороба Коновалова-Вільсона та ін.);
11) спадкові синдроми порушення всмоктування в травному тракті (муковісцидоз, непереносимість лактози та ін.)
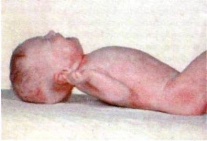 Фенілкетонурія (ФКУ) Аутосомно-рецесивний тип успадкування. ФКУ належить до спадкових порушень обміну, які широко відомі і досить детально вивчені. Фенілаланін (ФА) належить до незамінних амінокислот. Він надходить із продуктами харчування і не використовується для синтезу білка, а в нормі розпадається за тирозиновим шляхом. Метаболіти разом з надлишком фенілаланіну порушують процеси метаболізму і викликають ушкодження клітин головного мозку.
Фенілкетонурія (ФКУ) Аутосомно-рецесивний тип успадкування. ФКУ належить до спадкових порушень обміну, які широко відомі і досить детально вивчені. Фенілаланін (ФА) належить до незамінних амінокислот. Він надходить із продуктами харчування і не використовується для синтезу білка, а в нормі розпадається за тирозиновим шляхом. Метаболіти разом з надлишком фенілаланіну порушують процеси метаболізму і викликають ушкодження клітин головного мозку.
Дитина при народженні виглядає здоровою. Відставання у психічному розвитку може відбуватися поступово і стати очевидним тільки через кілька місяців. Ранній симптом захворювання – блювота. Можливі підвищена дратівливість, екзема і судоми.У старшому віці неліковані діти стають гіперактивними, здійснюють безцільні рухи, ритмічні погойдування. При об'єктивному обстеженні привертає увагу те, що дитина виглядає білявою, у неї світла шкіра і блакитні очі. У деяких хворих з'являється себорейна чи екзематозна шкірна висипка. Від них відходить незвичний запах фенілоцтової кислоти, який характеризують як запліснявілий, мишачий чи вовчий. У більшості дітей визначаються гіпертонус і підвищення глибоких сухожилкових рефлексів, тремор. Близько 1/4 дітей страждають судомами. Поведінка їх змінена: вони здаються або добродушними і привітними, або нервовими і запальними. Часто в нелікованих дітей визначають мікроцефалію, виступаючу верхню щелепу з широко розставленими зубами, гіпоплазією емалі, відставання в рості. У нелікованих випадках розвивається олігофренія різного ступеня, включно до ідіотії.
Для проведення якісних проб об'єктом дослідження є сеча дитини (тест Феллінга, або «пелюшкова проба»): на мокру пелюшку немовляти наноситься декілька крапель 10 % розчину FеСl3 і з появою зеленої плями діагностується ФКУ.
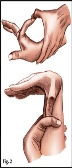
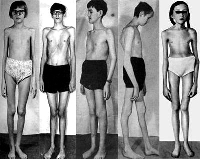 Синдром Марфана ("павукові пальці") Аутосомно-домінантний тип успадкування. Причина хвороби - мутація в гені, відповідальному за синтез білка сполучнотканинних волокон фібриліну. Блокування його синтезу призводить до підвищеної розтяжності сполучної тканини.
Синдром Марфана ("павукові пальці") Аутосомно-домінантний тип успадкування. Причина хвороби - мутація в гені, відповідальному за синтез білка сполучнотканинних волокон фібриліну. Блокування його синтезу призводить до підвищеної розтяжності сполучної тканини.
Хворих з синдромом Марфана відрізняють високий зріст, довгі павукоподібні пальці, деформація грудної клітини (воронкоподібна, килевидная, сплощена), плоскостопість. Нерідко мають місце стегнові та пахові грижі, гіпоплазія (недорозвинення) м'язів, м'язова гіпотонія, погіршення зору, зміна форми і розміру кришталика, значна міопія аж до відшарування сітківки, гетерохромія (різне фарбування ділянок райдужки); підвивих кришталика, катаракта, косоокість.
Крім перерахованого, при синдромі Марфана характерні вроджені вади серця, розширення аорти з розвитком аневризми. Нерідко відзначаються розлади органів дихання, ураження шлунково-кишкового тракту та сечовивідної системи.
 Лікування в основному симптоматичне. Позитивне дію роблять масаж, лікувальна фізкультура, а в ряді випадків оперативне втручання. Велике значення має рання діагностика захворювання. Частота синдрому Марфана в популяції дорівнює 1:10.0 (1:15.000).
Лікування в основному симптоматичне. Позитивне дію роблять масаж, лікувальна фізкультура, а в ряді випадків оперативне втручання. Велике значення має рання діагностика захворювання. Частота синдрому Марфана в популяції дорівнює 1:10.0 (1:15.000).
Синдромом Марфана страждали президент США Авраам Лінкольн, великий італійський скрипаль і композитор Ніколо Паганіні.
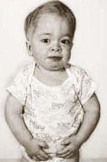
Хвороба Тея-Сакса. Пов'язана з обміном ліпідів. Це захворювання з аутосомно-рецесивним типом успадкування.
Гістологічно проявляється картина генералізованого розпаду гангліозних клітин нервової системи. Хвороба починається у віці 4-6 місяців. Часто це захворювання носить сімейний характер. У хворих рано виявляється зниження зору. Дитина не може фіксувати погляд, не стежить за іграшками. Досить рано на очному дні виявляється симптом "вишневої кісточки" - вишнево-червона цятка в макулярній ділянці, оточена сірувато-білим кільцем. Згодом розвиваються атрофія зорових нервів і повна сліпота. Зникають орієнтувальні і захисні реакції. Порушення призводять до повної нерухомості. При хворобі Тея - Сакса спостерігається симптом підвищеної реакції на звукові подразники - діти різко здригаються від звичайного звуку, можуть відзначатися судоми. Смерть настає в середньому через 1-2 роки після початку захворювання.
Фруктоземія - це спадкове захворювання, що передається за аутосомно-рецесивним типом, воно пов'язане з обміном вуглеводів. Поширеність його 1:20000 населення. Клініка фруктоземії нагадує галактоземію, але вона звичайно виникає, коли дитина починає одержувати соки, плоди й овочі або вживати цукор. Організм дитини не сприймає фрукти, у неї з'являється блювота, характерний розвиток гіпотрофії, гепатоспленомегалії, асциту. Вживання великої кількості фруктози може призвести до гострої гіпоглікемії, що супроводжується судомами, тремором і розвитком коматозного стану.
Діагностика фруктоземії полягає у виявленні фруктозурії (реакція Селіванова) при навантаженні фруктозою, гіпоглікемії, ознаках ураження печінки.
Лікування фруктоземії полягає у виключенні з раціону соків, ягід, фруктів і овочів. При своєчасному виключенні фруктози дитина розвивається нормально і прояви захворювання набувають зворотного розвитку.
Гемофілія – це спадкова патологія системи гемостазу, в основі якої лежить зниження або порушення синтезу VIII, IX або XI факторів згортання крові. Специфічним проявом гемофілії служить схильність організму дитини до різних кровотеч. Лікування гемофілії передбачає проведення замісної терапії: трансфузії гемоконцентратів з факторами згортання VIII або IX, свіжозамороженої плазми, антигемофільного глобуліну та ін.
Гени, що обумовлюють розвиток гемофілії, зчеплені зі статевою Х-хромосомою, тому захворювання успадковується за рецесивною ознакою по жіночій лінії. Спадковою гемофілію хворіють виключно особи чоловічої статі, а жінки є кондукторами (носіями) гена гемофілії, та передають захворювання своїм синам.
Тим не менше, відомі також поодинокі випадки гемофілії у дівчаток, народжених від матері-носія та хворого на гемофілію батька.
Вроджена гемофілія зустрічається майже у 70% пацієнтів. В цьому випадку успадковується форма та важкість гемофілії. Близько 30% спостережень припадає на спорадичні форми гемофілії, пов’язані з мутацією в локусі, що кодує синтез плазмових факторів згортання крові на Х-хромосомі. Надалі така спонтанна форма гемофілії стає спадковою.
Різновиди гемофілії
Залежно від дефіциту того чи іншого фактора згортання крові, розрізняють гемофілію А (класичну), В (хвороба Крістмаса), С та інші. Класична гемофілія буває в переважної більшості (близько 85%) дітей і пов’язана з дефіцитом VIII фактора згортання.
При спадковій гемофілії В, що становить 13% випадків, має місце недостаток IX фактора
Гемофілія С зустрічається з частотою 1-2% і зумовлена недостатністю XI фактора згортання крові (попередника тромбопластину). На інші різновиди гемофілії припадає менше 0,5% випадків.
Подружні пари, що знаходяться в групі ризику по народженню дитини з гемофілією, повинні пройти медико-генетичне консультування ще на етапі планування вагітності. Виявлення носіїв дефектного гена дозволяє аналізувати генеалогічні дані за допомогою молекулярно-генетичних досліджень. Можливе проведення пренатальної діагностики гемофілії за допомогою біопсії хоріона або амніоцентезу та дослідження ДНК клітинного матеріалу.
Повне позбавлення цього захворювання є неможливим, тому основу лікування становить замісна гемостатична терапія концентратами VIII і IX факторів згортання крові. Зокрема, через брак фінансування ліки закупаються на мінімальному рівні, тільки для оперативного реагування для дорослих хворих і для профілактики для новонароджених. При цьому ліки не можна купити в аптеці. Профілактичне лікування - це і є найбільш сучасний вид підтримки спочатку дітей, потім дорослих людей, щоб вони не ставали інвалідами, а вели нормальний спосіб життя. (Нині в Україні таких дітей всього 40)
Для прикладу, в Ізраїлі хворі на гемофілію навіть служать в армії.
Профілактика передбачає проведення медико-генетичного консультування подружніх пар, що мають обтяжений сімейний анамнез по гемофілії. Діти, хворі на гемофілію, завжди повинні мати при собі спеціальний паспорт, в якому вказано тип захворювання, група крові та Rh-приналежність.
Хромосомні хвороби, обумовлені зміною числа аутосом .
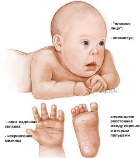 Синдром Дауна (трисомія 21). Клінічну картину синдрому вперше в 1866 р. описав
Синдром Дауна (трисомія 21). Клінічну картину синдрому вперше в 1866 р. описав  англійський лікар Л. Даун. У 1959 р. французький учений І.Лежен виявив у каріотипі хворих зайву хромосому 21. Каріотипи хворих – 47, ХХ, 21+ або 47, ХY, 21+. Частота 1:1100, а в деяких регіонах – 1:700-1:800 новонароджених. Ризик народження дітей з синдромом Дауна зростає з віком матері. На частоту їх народження не впливають статеві, расові, географічні і популяційні відмінності.
англійський лікар Л. Даун. У 1959 р. французький учений І.Лежен виявив у каріотипі хворих зайву хромосому 21. Каріотипи хворих – 47, ХХ, 21+ або 47, ХY, 21+. Частота 1:1100, а в деяких регіонах – 1:700-1:800 новонароджених. Ризик народження дітей з синдромом Дауна зростає з віком матері. На частоту їх народження не впливають статеві, расові, географічні і популяційні відмінності.
Клінічні діагностичні ознаки:
— низький зріст;
— різні ступені розумової відсталості;
— черепно-лицеві аномалії: косий розріз очей, коротка шия, плоске обличчя, маленький короткий ніс, великий язик, маленькі деформовані вуха;
— м’язова гіпотонія;
— розхитаність суглобів;
— поперечна складка на долонях;
— клинодактилія мізинця;
— вроджені вади внутрішніх органів (серця).
Знижений імунітет часто є причиною смерті цих дітей.
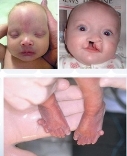 Синдром Патау (трисомія-13). Каріотип 47, ХХ, 13+ або 47, ХY, 13+. Частота 1:5000 — 1:7000 новонароджених.
Синдром Патау (трисомія-13). Каріотип 47, ХХ, 13+ або 47, ХY, 13+. Частота 1:5000 — 1:7000 новонароджених.
Клінічні діагностичні ознаки:
— щілини верхньої губи і піднебіння,
— зменшений об’єм черепа,
— перекошений, низький лоб,
— маленькі очі,
— відсутність одного або обох очних яблук,
— перенісся запале,
— деформовані вушні раковини, полідактилія;
— вроджені вади серця, інших внутрішніх органів.
Більшість дітей вмирає в перші тижні або місяці. Вирішальним у діагностиці є цитогенетичне дослідження.
Хромосомні хвороби, обумовлені зміною числа статевих хромосом
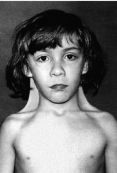 Синдром Шерешевського-Тернера. Каріотип 45,Х0. У клітинах відсутні тільця статевого хроматину. Частота 1:2000-1:5000. Поряд із справжньою моносомією можуть зустрічатись делеції плеч (довгого або короткого), кільцеві хромосоми. Синдром описали російський клініцист М.А. Шерешевський (1925) і Г.Тернер (1938).
Синдром Шерешевського-Тернера. Каріотип 45,Х0. У клітинах відсутні тільця статевого хроматину. Частота 1:2000-1:5000. Поряд із справжньою моносомією можуть зустрічатись делеції плеч (довгого або короткого), кільцеві хромосоми. Синдром описали російський клініцист М.А. Шерешевський (1925) і Г.Тернер (1938).
Клінічні діагностичні ознаки:
— синдром виявляється у жінок;
— низький зріст,
— коротка шия з надлишком шкіри і крилоподібними складками (шия сфінкса),
— низька межа росту волосся на потилиці,
— грудна клітка щитоподібної форми з широко розставленими сосками, дисгенезія гонад.
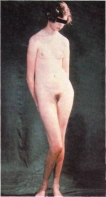 Інтелектуальний розвиток хворих із синдромом Шерешевського - Тернера в більшості випадків нормальний або близький до норми. Хворим властиві деякі риси недорозвинення емоційно-вольової сфери, вони вередливі, вперті й у той же час легко піддаються впливу, часто ейфоричні.
Інтелектуальний розвиток хворих із синдромом Шерешевського - Тернера в більшості випадків нормальний або близький до норми. Хворим властиві деякі риси недорозвинення емоційно-вольової сфери, вони вередливі, вперті й у той же час легко піддаються впливу, часто ейфоричні.
Синдром трипло-Х (синдром «супержінки»). Каріотип 47, ХХХ. Переважна більшість таких жінок мають нормальний фізичний та розумовий розвиток і виявляються випадково при обстеженні. Лише в деяких з них порушена репродуктивна функція. Більшість жінок мають нормальну плодючість, хоча зростає ризик мимовільних викиднів і хромосомних аберацій у нащадків. У клітинах – по два тільця статевого хроматину. При збільшенні числа Х-хромосом наростає ступінь відхилення від норми. У жінок з тетра- і пентасомією описані розумова відсталість, черепно-лицеві дизморфії, аномалії зубів, скелета і статевих органів. Однак жінки навіть з тетрасомією по Х- хромосомі мають нащадків.
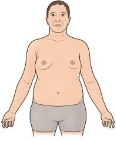 Синдром Клайнфельтера. Каріотип 47, ХХY. Частота 1:400. Синдром виявляється лише в осіб чоловічої статі переважно при статевому дозріванні.
Синдром Клайнфельтера. Каріотип 47, ХХY. Частота 1:400. Синдром виявляється лише в осіб чоловічої статі переважно при статевому дозріванні.
Клінічні діагностичні ознаки:
— високий зріст,
— довгі кінцівки,
— збільшення молочних залоз,
— відсутність сперматогенезу,
— недорозвинення статевих залоз.
Інколи хворі з синдромом Клайнфельтера мають 48 і 49 хромосом (48, ХХXY; 49, ХХХХY). Чим більше Х-хромосом у каріотипі, тим вища ймовірність розвитку розумової відсталості.
 Синдром дисомії за Y-хромосомою (синдром «суперчоловіка»). Каріотип 47, ХYY. Частота 1:1000. Синдром виявляється в осіб чоловічої статі. За своїм розумовим і фізичним розвитком такі чоловіки не відрізняються від нормальних осіб. Помітних відхилень у статевому і гормональному статусі не виявлено. Проте деякі клініцисти вказували на підвищений ступінь агресивності в окремих з них. Чоловіки, у каріотипах яких присутні зайві Y-хромосоми, також рідко звертаються до фахівців і навіть не знають про свою особливість. Однак з’ясувалася цікава закономірність: кількість носіїв зайвої Y-хромосоми серед ув’язнених істотно перевищує кількість таких людей у середньому по людській популяції. Виявилося, що наявність у каріотипі зайвої Y-хромосоми провокує в чоловіка агресивність. Зовні такі хворі — високі чоловіки з надто довгими руками і лютим виразом обличчя.
Синдром дисомії за Y-хромосомою (синдром «суперчоловіка»). Каріотип 47, ХYY. Частота 1:1000. Синдром виявляється в осіб чоловічої статі. За своїм розумовим і фізичним розвитком такі чоловіки не відрізняються від нормальних осіб. Помітних відхилень у статевому і гормональному статусі не виявлено. Проте деякі клініцисти вказували на підвищений ступінь агресивності в окремих з них. Чоловіки, у каріотипах яких присутні зайві Y-хромосоми, також рідко звертаються до фахівців і навіть не знають про свою особливість. Однак з’ясувалася цікава закономірність: кількість носіїв зайвої Y-хромосоми серед ув’язнених істотно перевищує кількість таких людей у середньому по людській популяції. Виявилося, що наявність у каріотипі зайвої Y-хромосоми провокує в чоловіка агресивність. Зовні такі хворі — високі чоловіки з надто довгими руками і лютим виразом обличчя.
За високий зріст та асоціальну поведінку носіїв зайвої Y-хромосоми у середовищі генетиків іноді називають «баскетбольною командою в’язниць».
Хромосомні хвороби, зумовлені порушенням структури хромосом.
 Синдром «Котячого крику» — клінічні прояви даного синдрому полягає в порушенні будови гортані, нявкаючий тембр голосу, розумова відсталість. Викликане делецією (відрив хромосоми) 5р.
Синдром «Котячого крику» — клінічні прояви даного синдрому полягає в порушенні будови гортані, нявкаючий тембр голосу, розумова відсталість. Викликане делецією (відрив хромосоми) 5р.
Найбільш характернина ознака – «крик кішки» – обумовлена змінами гортані (звуження, м’якість хрящів, зменшеня надгортанника, незвична складчастість слизової оболонки). Практично у всіх хворих є зміни мозкового черепа та обличчя: місяцеподібне обличчя, мікроцефалія, антимонголоїдний розріз очей, високе піднебіння, плоска спинка носа. Вушні раковини розмішені низько та деформовані. Крім того, зустрічаються вроджені вади серця та інших внутрішніх органів, опорно-рухового апарату (синдактилія стоп, косолапість). Харакрерна м’язева гіпотонія.
Більшість хворих помирає в перші роки, тільки 10 % досягають 10 річного віку. У всіх випадках хворим та їхнім батькам показане цитогенетичне обстеження. Хворіти можуть люди чоловічої і жіночої статі.
Мультифакторіальні (полігенні) захворювання - це захворювання зі спадковою схильністю, у генезі яких поєднуються взаємодія спадкових і зовнішніх факторів. Мультифакторіальні захворювання - це такі патологічні стани, для прояву яких необхідні дві умови:
1. Наявність спадкової схильності.
2. Несприятливі впливи зовнішнього середовища.
З певною часткою умовності мультифакторіальні хвороби можна розділити на:
1) вроджені вади розвитку,
2) поширені психічні і нервові хвороби,
3) поширені хвороби «середнього» віку.


про публікацію авторської розробки
Додати розробку