Презентація до уроку з біології "Генні захворювання"
Генні захворювання – це велика група захворювань, які виникають в результаті пошкодження ДНК на рівні гену.
Загальна частота хвороб в популяції складає 1-2 %.
Більшість генних патологій обумовлена мутаціями в структурних генах, які функціонують через синтез поліпептидів – білків. Будь-яка мутація гену приводить до зміни структури або кількості білків. В даній розробці описана класифікація геннних захворювань ВООЗ.


Генні захворювання

Генні захворювання – це велика група захворювань, які виникають в результаті пошкодження ДНК на рівні гену. Загальна частота хвороб в популяції складає 1-2 %. Більшість генних патологій обумовлена мутаціями в структурних генах, які функціонують через синтез поліпептидів – білків. Будь-яка мутація гену приводить до зміни структури або кількості білків.
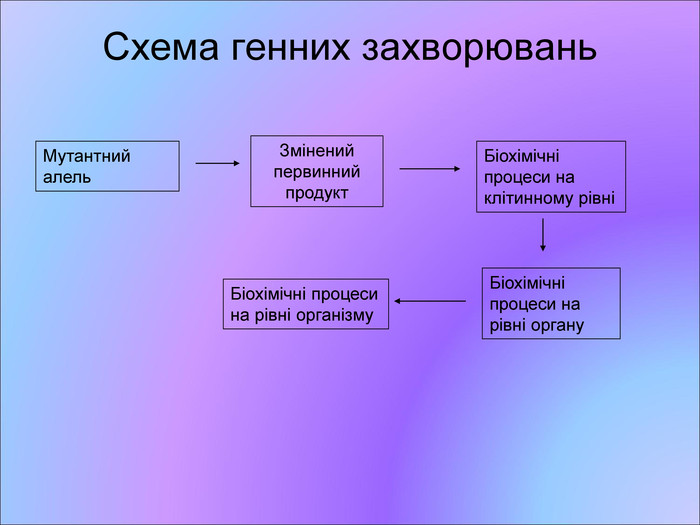
Схема генних захворювань Мутантний алель Змінений первинний продукт Біохімічні процеси на клітинному рівні Біохімічні процеси на рівні органу Біохімічні процеси на рівні організму
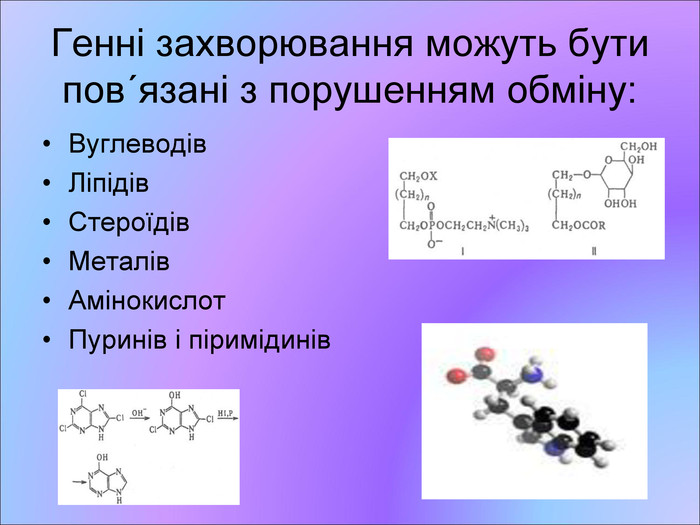
Генні захворювання можуть бути повґязані з порушенням обміну: Вуглеводів Ліпідів Стероїдів Металів Амінокислот Пуринів і піримідинів
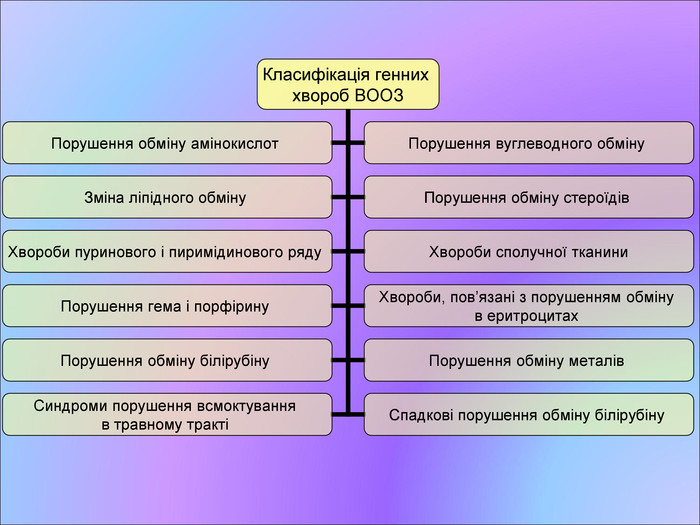
Класифікація генних хвороб ВООЗ Порушення обміну амінокислот Порушення вуглеводного обміну Зміна ліпідного обміну Порушення обміну стероїдів Хвороби пуринового і пиримідинового ряду Хвороби сполучної тканини Порушення гема і порфірину Хвороби, пов’язані з порушенням обміну в еритроцитах Порушення обміну білірубіну Порушення обміну металів Синдроми порушення всмоктування в травному тракті Спадкові порушення обміну білірубіну

Спадкові хвороби амінокислотного обміну Це – найчисельніша група спадкових захворювань обміну речовин. Майже всі вони успадковуються за аутосомно-рецесивним типом. Причиною захворювань є недостатність того чи іншого ферменту, який відповідає за синтез амінокислоти. До спадкових захворювань з порушенням амінокислотного обміну належать: фенілкетонурія, альбінізм та ін.
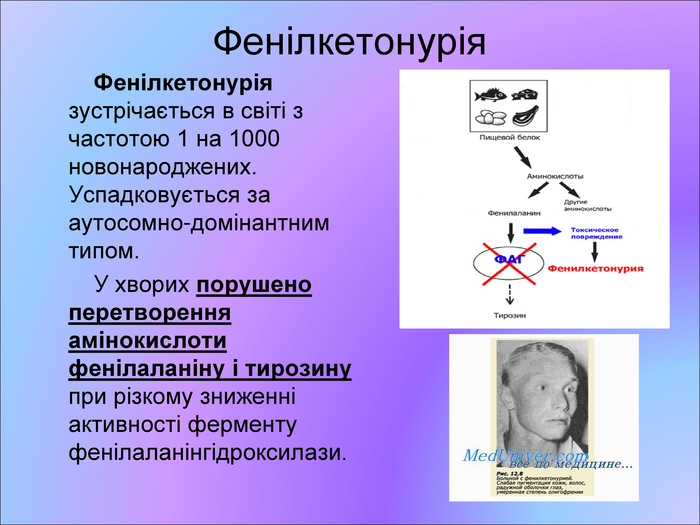
Фенілкетонурія Фенілкетонурія зустрічається в світі з частотою 1 на 1000 новонароджених. Успадковується за аутосомно-домінантним типом. У хворих порушено перетворення амінокислоти фенілаланіну і тирозину при різкому зниженні активності ферменту фенілаланінгідроксилази.

Симптоматика захворювання Підвищена збудливість Судоми Схильність до дерматитів Сеча і піт мають характерний “мишачий” запах Судомні припадки Олігофренія

Альбінізм (очно-шкірний) Альбінізм зустрічається з частотою 1 на 39000, успадковується за аутосомно-рецесивним типом. Ген локалізований на довгому плечі 12 хромосоми. Хвороба обумовлена відсутність синтезу ферменту тирозинази.
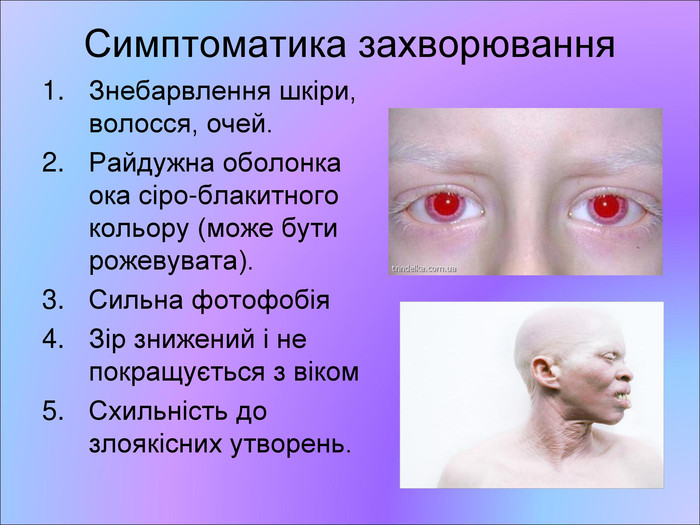
Симптоматика захворювання Знебарвлення шкіри, волосся, очей. Райдужна оболонка ока сіро-блакитного кольору (може бути рожевувата). Сильна фотофобія Зір знижений і не покращується з віком Схильність до злоякісних утворень.

Спадкові захворювання, пов’язані з порушенням обміну вуглеводів Відомо, що вуглеводи входять до складу ряду біологічно-активних речовин – гормонів, ферментів, мукополісахаридів, які виконують енергетичну та структурну функції. Внаслідок порушення вуглеводного обміну розвивається глікогенова хвороба, галактоземія.
Глікогенова хвороба Глікогенова хвороба пов’язана з порушенням синтезу та розкладу глікогену – тваринного крохмалю.
Хвороба Гірке Глікогеноз І типу – хвороба Гірке – у хворих в печінці, нирках та слизовій оболонці кишечнику накопичується велика кількість глікогену, тому що перетворення глікогену у глюкозу не відбувається, так як відсутній фермент глюко-6-фосфатаза, який регулює кількість глюкози у крові. Хвороба Гірке успадковується за аутосомно-рецесивним типом. Глікоген Глюкоза
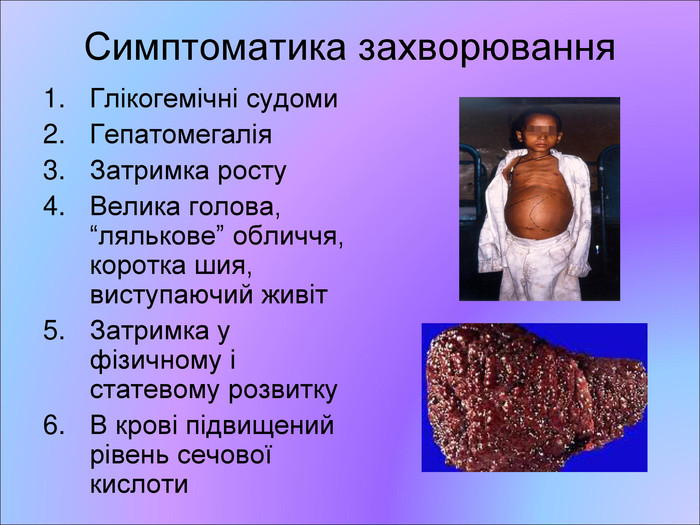
Симптоматика захворювання Глікогемічні судоми Гепатомегалія Затримка росту Велика голова, “лялькове” обличчя, коротка шия, виступаючий живіт Затримка у фізичному і статевому розвитку В крові підвищений рівень сечової кислоти

Хвороба Помпе Глікогеноз ІІ ступеня – хвороба Помпе – протікає в складній формі, глікоген накопичується в печінці, в скелетних м’язах, міокарді, легенях, селезінці, наднирниках, стінках судин, в нейронах. Симптоматика: 1. Мґязова слабкість 2. Кардіомегалія 3. Макроглоссія 4. Важкі форми пневмонії

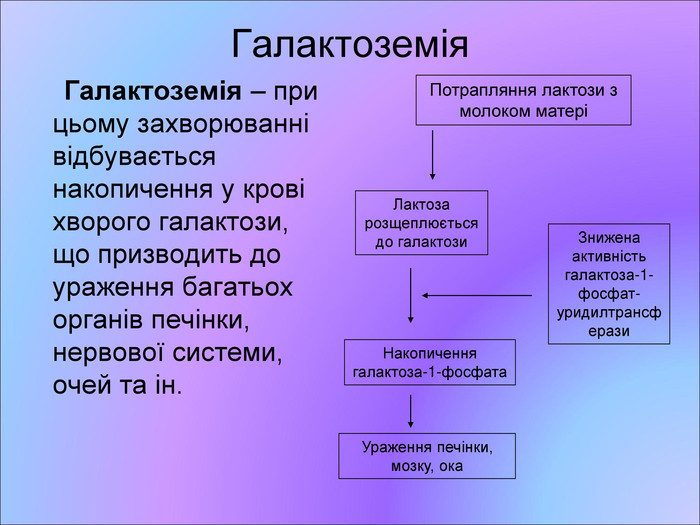
Галактоземія Галактоземія – при цьому захворюванні відбувається накопичення у крові хворого галактози, що призводить до ураження багатьох органів печінки, нервової системи, очей та ін. Потрапляння лактози з молоком матері Лактоза розщеплюється до галактози Знижена активність галактоза-1-фосфат-уридилтрансферази Накопичення галактоза-1-фосфата Ураження печінки, мозку, ока

Симптоматика захворювання Розлади травлення Інтоксикація Гепатомегалія, печінкова недостатність, жовтяниця Катаракта Розумова відсталість

Порушення ліпідного обміну Спадкові захворювання обміну ліпідів (ліпідози) розрізняються на 2 основних типи: 1) внутріклітинні, при яких відбувається накопичення ліпідів в клітинах різних тканин; 2) хвороби з порушенням метаболізму ліпопротеїдів, які містяться в крові. До найбільш вивчених захворювань ліпідного обміну першого типу відносять хворобу Гоше, хворобу Німанна-Піка та амавротичну ідіотію (хворобу Тея-Сакса).
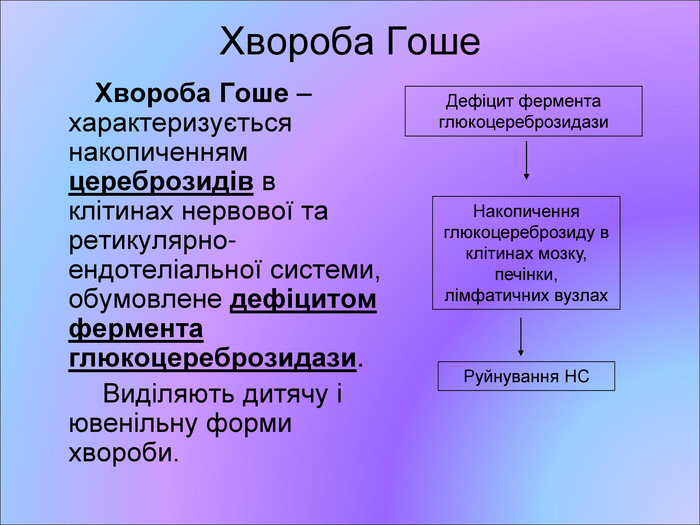
Хвороба Гоше Хвороба Гоше – характеризується накопиченням цереброзидів в клітинах нервової та ретикулярно-ендотеліальної системи, обумовлене дефіцитом фермента глюкоцереброзидази. Виділяють дитячу і ювенільну форми хвороби. Дефіцит фермента глюкоцереброзидази Накопичення глюкоцереброзиду в клітинах мозку, печінки, лімфатичних вузлах Руйнування НС

Симптоматика захворювання Дитяча форма хвороби Затримка розумового і фізичного розвитку Збільшення живота, печінки і селезінки Утруднення ковтання Спазм гортані Дихальна недостатність Судоми Ювенільна форма хвороби Пігментація шкіри Остеопороз Переломи Деформація кісток
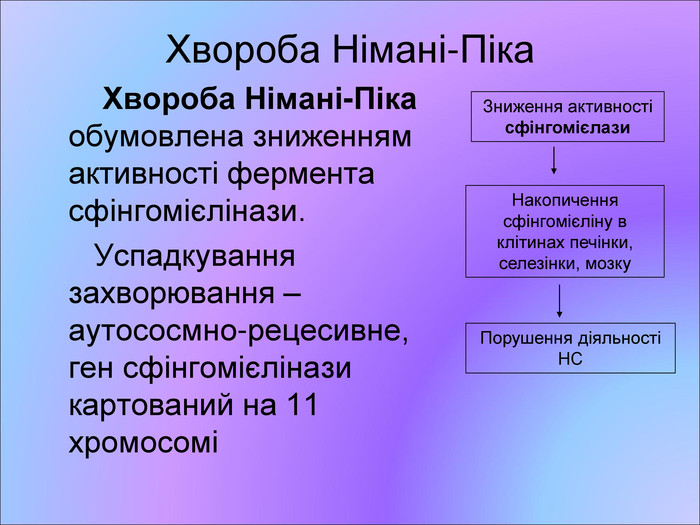
Хвороба Німані-Піка Хвороба Німані-Піка обумовлена зниженням активності фермента сфінгомієлінази. Успадкування захворювання – аутососмно-рецесивне, ген сфінгомієлінази картований на 11 хромосомі Зниження активності сфінгомієлази Накопичення сфінгомієліну в клітинах печінки, селезінки, мозку Порушення діяльності НС
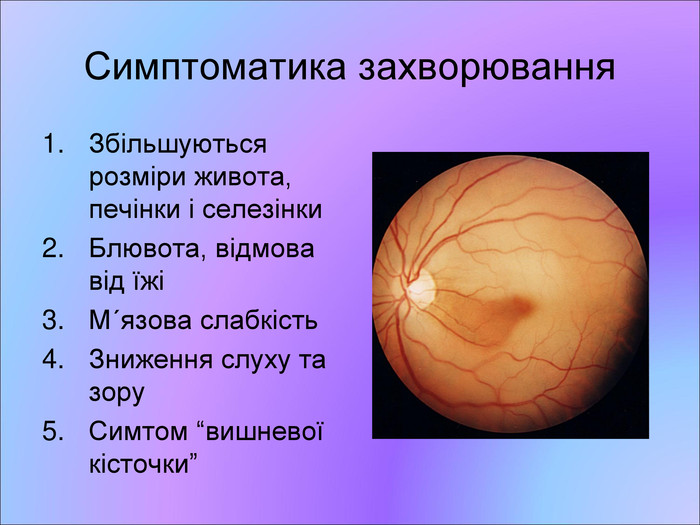
Симптоматика захворювання Збільшуються розміри живота, печінки і селезінки Блювота, відмова від їжі Мґязова слабкість Зниження слуху та зору Симтом “вишневої кісточки”
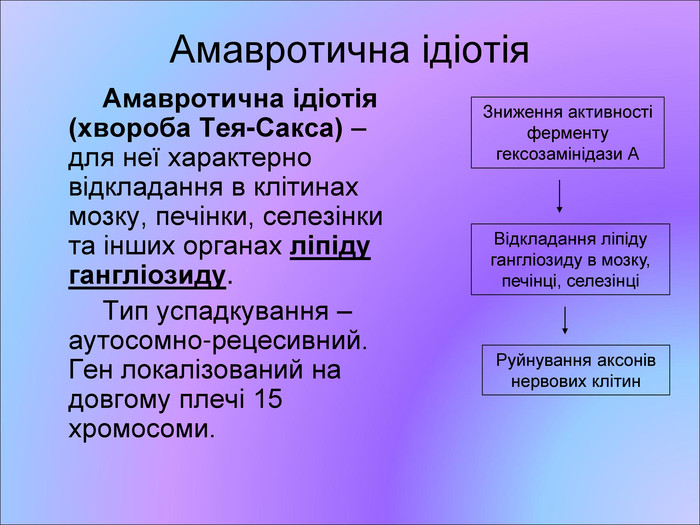
Амавротична ідіотія Амавротична ідіотія (хвороба Тея-Сакса) – для неї характерно відкладання в клітинах мозку, печінки, селезінки та інших органах ліпіду гангліозиду. Тип успадкування – аутосомно-рецесивний. Ген локалізований на довгому плечі 15 хромосоми. Зниження активності ферменту гексозамінідази А Відкладання ліпіду гангліозиду в мозку, печінці, селезінці Руйнування аксонів нервових клітин

Симптоматика захворювання 1. Дитина стає вґялою, малорухливою, байдужою 2. Затримка психічного розвитку 3. Судоми 4. Симптом “вишневої кісточки” 5. Сліпота, повне зневоднення.

Спадкові хвороби сполучної тканини Складна структура сполучної тканини склалась генетично. Патологія в її системі є причиною різних спадкових захворювань та обумовлена порушеннями структурних білків – колагенів. Більшість хвороб сполучної тканини пов’язано з дефектами опорно-рухового апарату та шкіри. До їх числа відносяться синдром Марфана, синдром Елерса-Данлоса, а також мукополісахаридози.
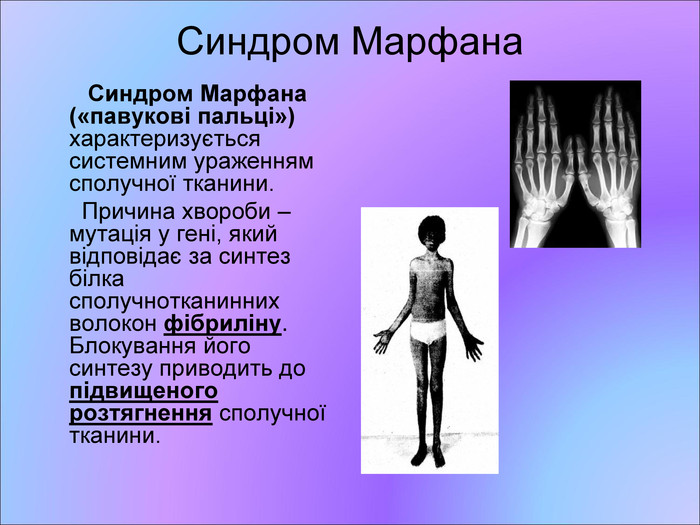
Синдром Марфана Синдром Марфана («павукові пальці») характеризується системним ураженням сполучної тканини. Причина хвороби – мутація у гені, який відповідає за синтез білка сполучнотканинних волокон фібриліну. Блокування його синтезу приводить до підвищеного розтягнення сполучної тканини.
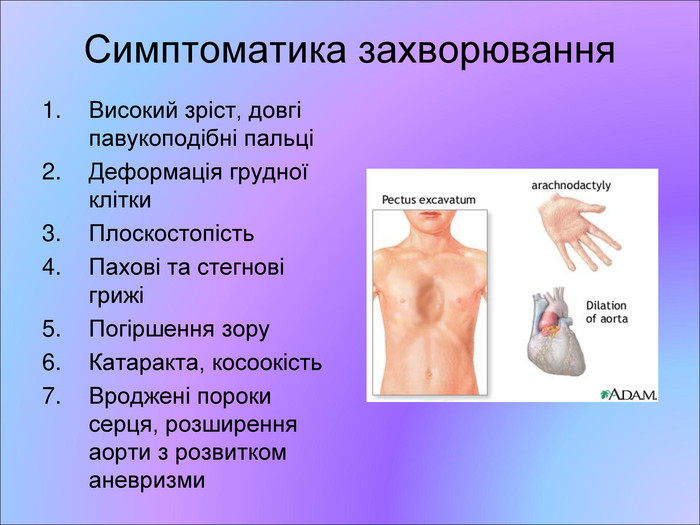
Симптоматика захворювання Високий зріст, довгі павукоподібні пальці Деформація грудної клітки Плоскостопість Пахові та стегнові грижі Погіршення зору Катаракта, косоокість Вроджені пороки серця, розширення аорти з розвитком аневризми
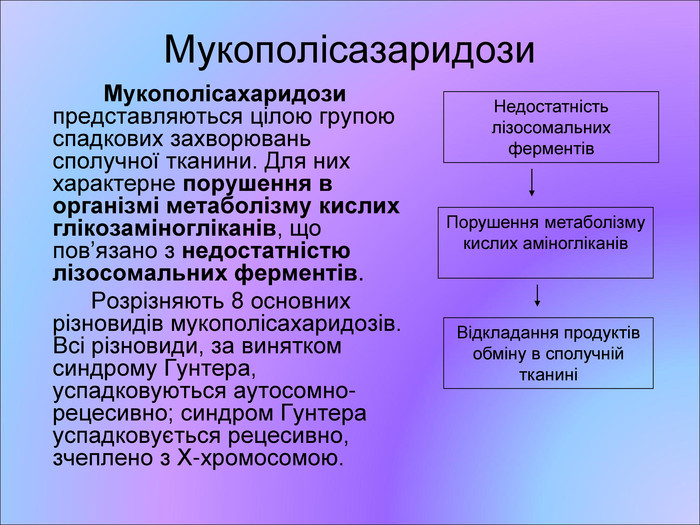
Мукополісазаридози Мукополісахаридози представляються цілою групою спадкових захворювань сполучної тканини. Для них характерне порушення в організмі метаболізму кислих глікозаміногліканів, що пов’язано з недостатністю лізосомальних ферментів. Розрізняють 8 основних різновидів мукополісахаридозів. Всі різновиди, за винятком синдрому Гунтера, успадковуються аутосомно-рецесивно; синдром Гунтера успадковується рецесивно, зчеплено з Х-хромосомою. Недостатність лізосомальних ферментів Порушення метаболізму кислих аміногліканів Відкладання продуктів обміну в сполучній тканині
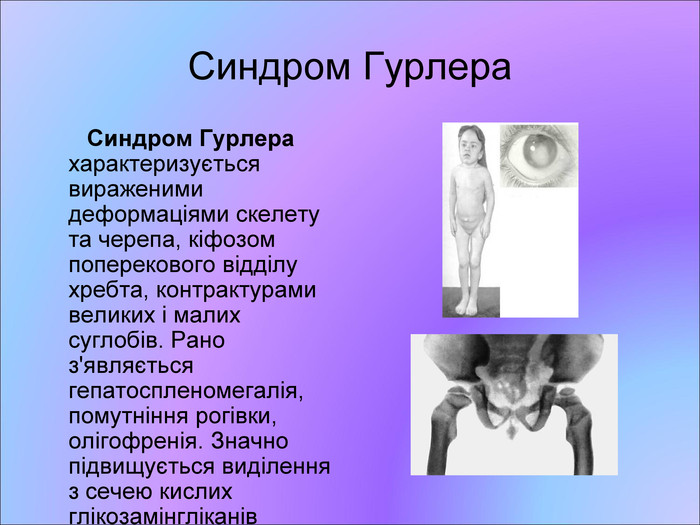
Синдром Гурлера Синдром Гурлера характеризується вираженими деформаціями скелету та черепа, кіфозом поперекового відділу хребта, контрактурами великих і малих суглобів. Рано з'являється гепатоспленомегалія, помутніння рогівки, олігофренія. Значно підвищується виділення з сечею кислих глікозамінгліканів

Синдром Санфіліппо та Моркіо Синдром Санфіліппо характеризується затримкою розумового розвитку та мовних функцій, симптомами внутрішньочерепної гіпертензії, менш вираженими кістково-суглобовими деформаціями, помутніння рогівки не спостерігається Синдром Моркіо проявляється вираженими деформаціями скелету, особливо грудної клітини, інші симптоми незначні або взагалі відсутні. Інтелект не змінюється. Виявляється підвищене виведення мукополісахаридних кератосульфатів з сечею
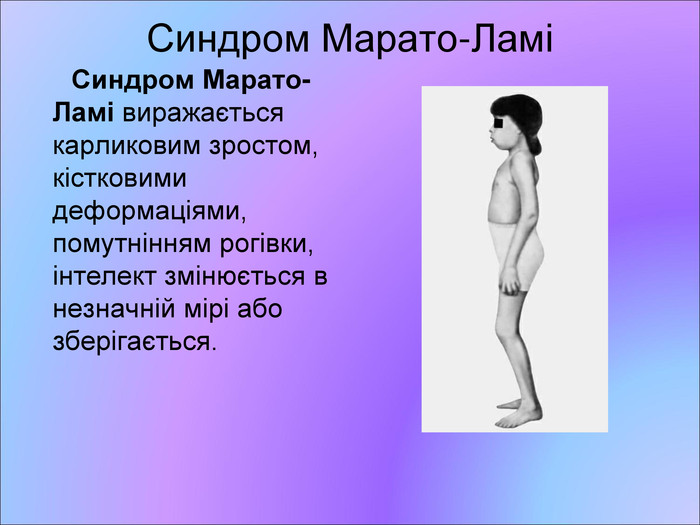
Синдром Марато-Ламі Синдром Марато-Ламі виражається карликовим зростом, кістковими деформаціями, помутнінням рогівки, інтелект змінюється в незначній мірі або зберігається.
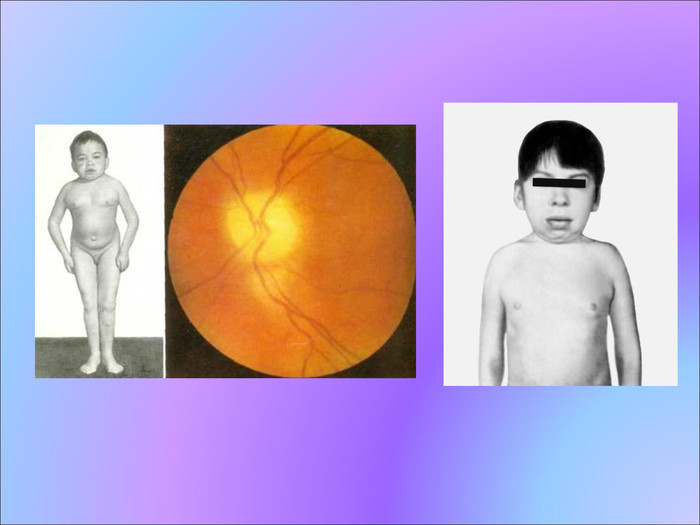
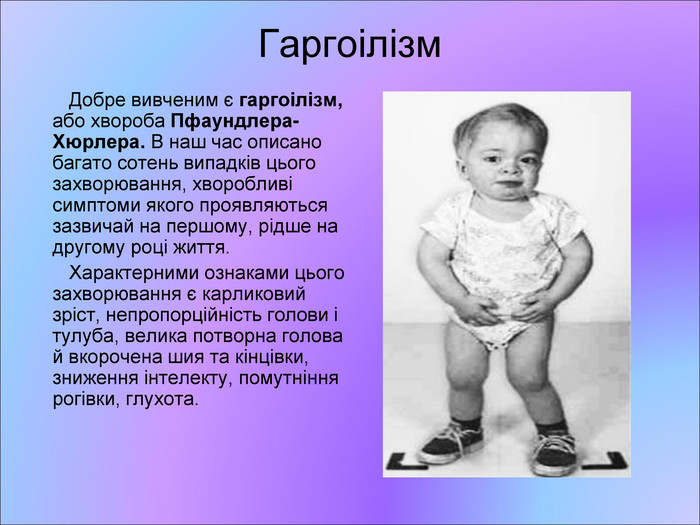
Гаргоілізм Добре вивченим є гаргоілізм, або хвороба Пфаундлера-Хюрлера. В наш час описано багато сотень випадків цього захворювання, хворобливі симптоми якого проявляються зазвичай на першому, рідше на другому році життя. Характерними ознаками цього захворювання є карликовий зріст, непропорційність голови і тулуба, велика потворна голова й вкорочена шия та кінцівки, зниження інтелекту, помутніння рогівки, глухота.
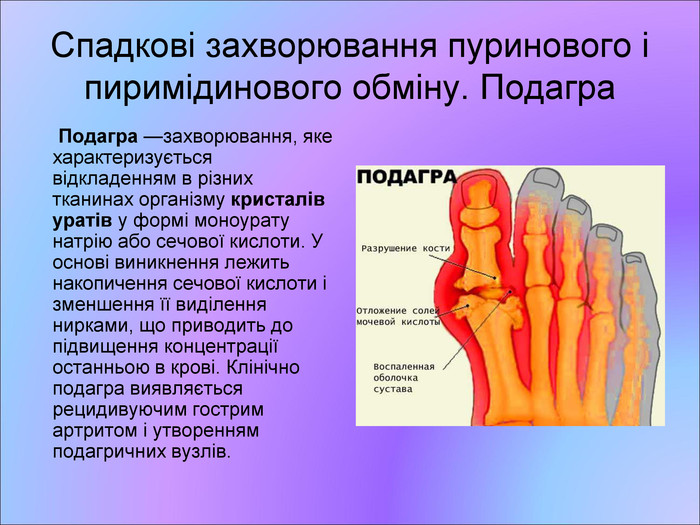
Спадкові захворювання пуринового і пиримідинового обміну. Подагра Подагра —захворювання, яке характеризується відкладенням в різних тканинах організму кристалів уратів у формі моноурату натрію або сечової кислоти. У основі виникнення лежить накопичення сечової кислоти і зменшення її виділення нирками, що приводить до підвищення концентрації останньою в крові. Клінічно подагра виявляється рецидивуючим гострим артритом і утворенням подагричних вузлів.
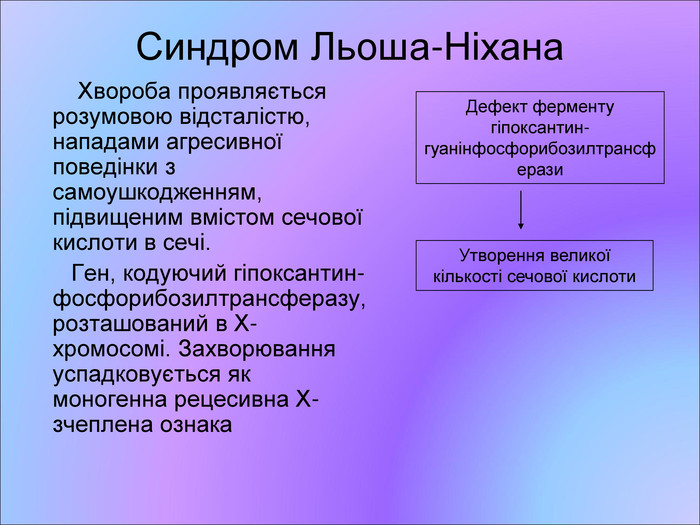
Синдром Льоша-Ніхана Хвороба проявляється розумовою відсталістю, нападами агресивної поведінки з самоушкодженням, підвищеним вмістом сечової кислоти в сечі. Ген, кодуючий гіпоксантин-фосфорибозилтрансферазу, розташований в Х-хромосомі. Захворювання успадковується як моногенна рецесивна Х-зчеплена ознака Дефект ферменту гіпоксантин-гуанінфосфорибозилтрансферази Утворення великої кількості сечової кислоти
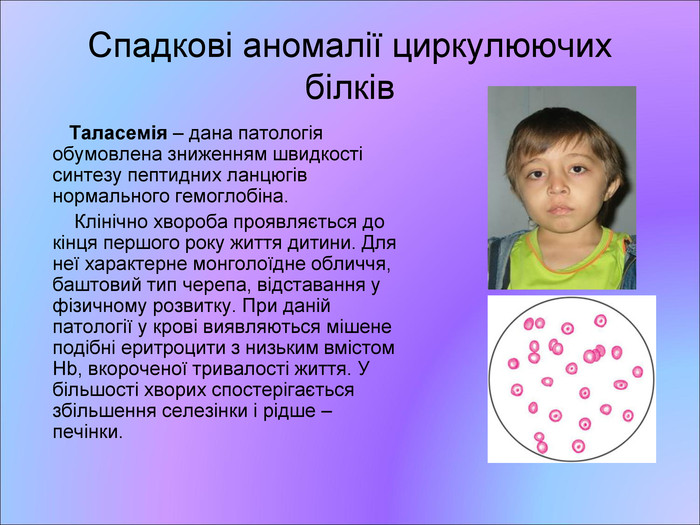
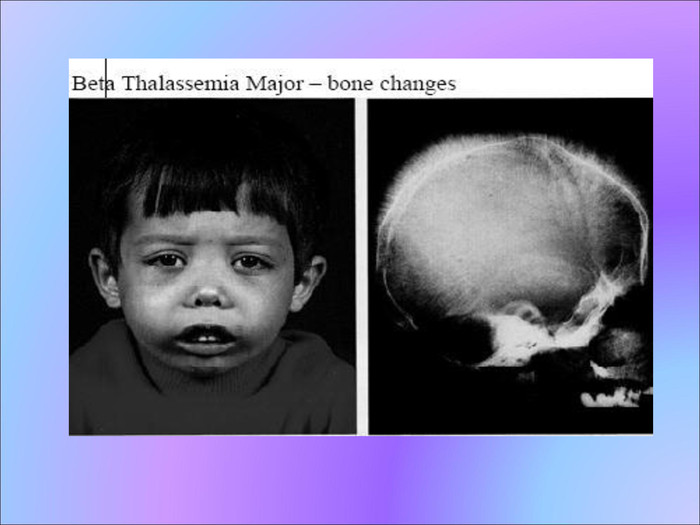
Спадкові аномалії циркулюючих білків Таласемія – дана патологія обумовлена зниженням швидкості синтезу пептидних ланцюгів нормального гемоглобіна. Клінічно хвороба проявляється до кінця першого року життя дитини. Для неї характерне монголоїдне обличчя, баштовий тип черепа, відставання у фізичному розвитку. При даній патології у крові виявляються мішене подібні еритроцити з низьким вмістом Hb, вкороченої тривалості життя. У більшості хворих спостерігається збільшення селезінки і рідше – печінки.
Хвороби, пов’язані з порушеннями обміну в еритроцитах Гемолітична анемія — групова назва достатньо рідких захворювань, , загальною ознакою яких є посилене руйнування еритроцитів, яке обумовлює, з одного боку, анемію і підвищене утворення продуктів розпаду еритроцитів, з іншого боку — реактивно посилений еритропоез.

Порушення обміну металів. Хвороба Вільсона-Коновалова Хвороба Вільсона — Коновалова або гепатоцеребральная дистрофія - вроджене порушення метаболізму міді, що приводить до важких спадкових хвороб центральної нервової системи і внутрішніх органів. Захворювання передається за аутосомно-рецесивним типом, обумовлено низьким або аномальним синтезом церулоплазміну — білка, що транспортує мідь.

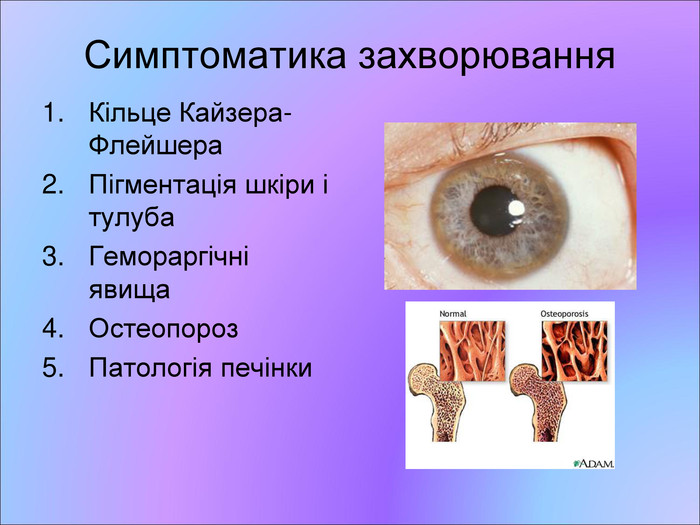
Симптоматика захворювання Кільце Кайзера-Флейшера Пігментація шкіри і тулуба Гемораргічні явища Остеопороз Патологія печінки
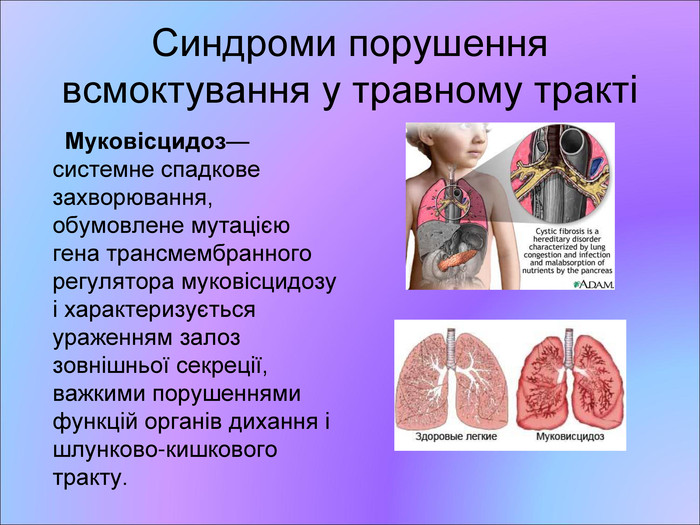
Синдроми порушення всмоктування у травному тракті Муковісцидоз— системне спадкове захворювання, обумовлене мутацією гена трансмембранного регулятора муковісцидозу і характеризується ураженням залоз зовнішньої секреції, важкими порушеннями функцій органів дихання і шлунково-кишкового тракту.

Симптоматика захворювання Згущування секретів залоз зовнішньої секреції Зміна фізико-хімічного складу секрету Меконієва непрохідність Зміни у органах дихання, часто – пневмонії.

Дякую за увагу!


про публікацію авторської розробки
Додати розробку