Презентація "Спадкові хвороби"


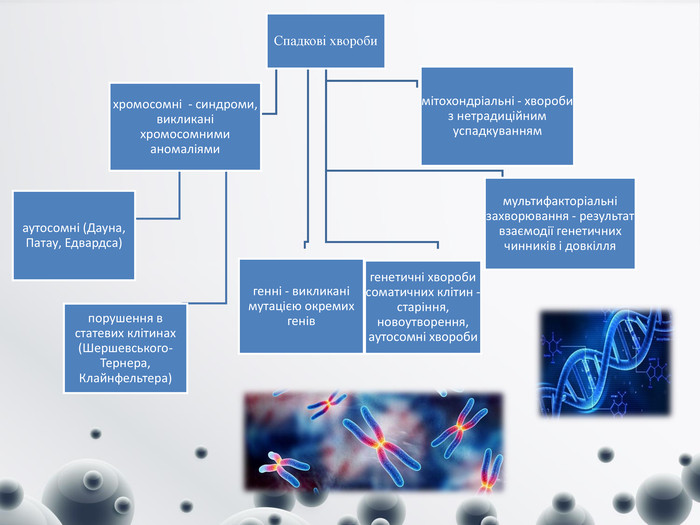
Спадкові хвороби

Синдром Дауна
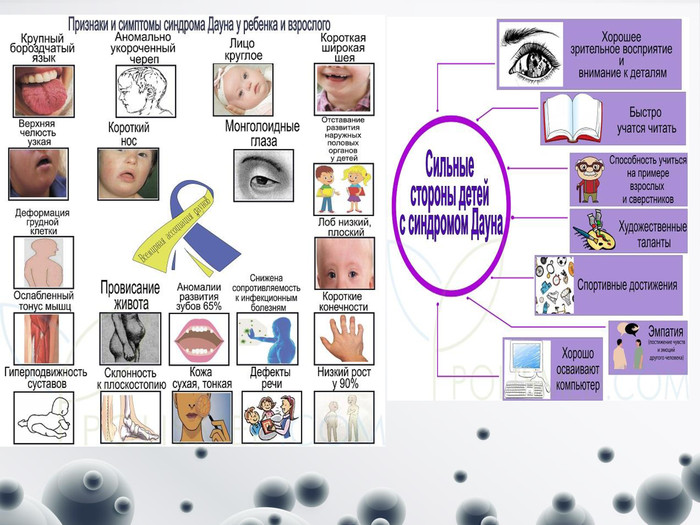

Синдром Дауна Генетична аномалія, викликана присутністю додаткової хромосомою в 21 парі- цілковита (трисомія 21) або часткова (внаслідок транслокації). Частота народження дітей з цією хворобою 1 на 1000. Великий ризик народження в родині , де чоловікові понад 50 років, а жінці за 40. Такі діти мають 47 хромосом у каріотипі замість звичних 46. Через це в них характерна зовнішність, повільніше розвиваються. Клінічні діагностичні ознаки: - низький зріст; - різні ступені розумової відсталості; - це специфічні риси обличчя- монголоїдний тип обличчя: косий розріз очей, коротка шия, епікант, плоске обличчя, маленький короткий ніс, великий язик, маленькі деформовані вуха; - м’язова гіпотонія; - розхитаність суглобів; - поперечна складка на долонях; - клинодактилія мізинця.; - вроджені вади внутрішніх органів (серця). Знижений імунітет часто є причиною смерті цих дітей.
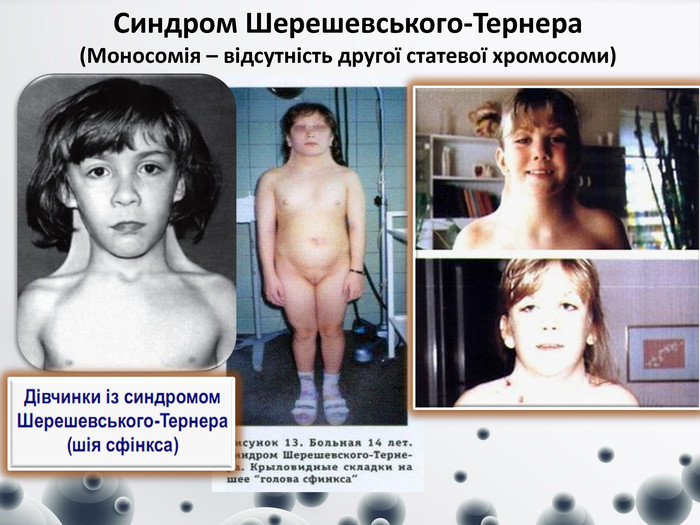
Синдром Шерешевського-Тернера (Моносомія – відсутність другої статевої хромосоми)
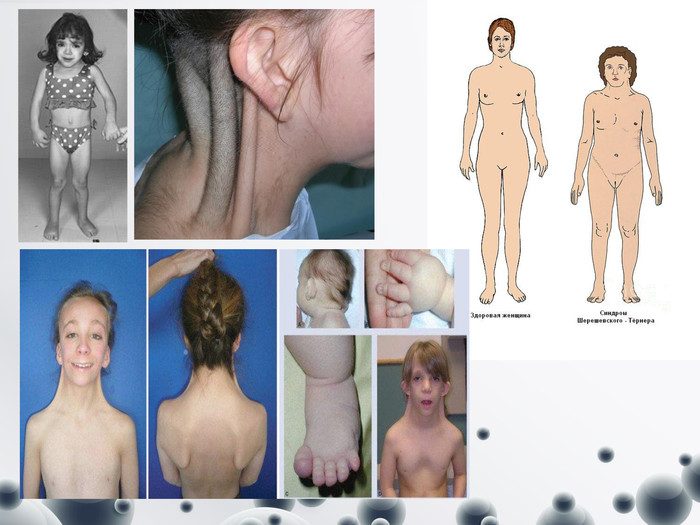

Синдром Шерешевського-Тернера (Моносомія – відсутність другої статевої хромосоми) У клітинах відсутні тільця статевого хроматину. Частота 1:2000-1:5000. Поряд із справжньою моносомією можуть зустрічатись делеції плеч (довгого або короткого), кільцеві хромосоми. Синдром описали російський клініцист М. А. Шерешевський (1925) і Г. Тернер (1938). Клінічні діагностичні ознаки: - синдром виявляється у жінок; - низький зріст, - коротка шия з надлишком шкіри і крилоподібними складками (шия сфінкса), - низька межа росту волосся на потилиці, - грудна клітка щитоподібної форми з широко розставленими сосками, дисгенезія гонад.- недорозвиненість нижньої щелепи- низькорозташовані деформовані вуха- високе тверде піднебіння - косоокість, катаракта - дефект слуху- аномалія сечової системи (подвооєння нирок, сечовивідних шляхів)
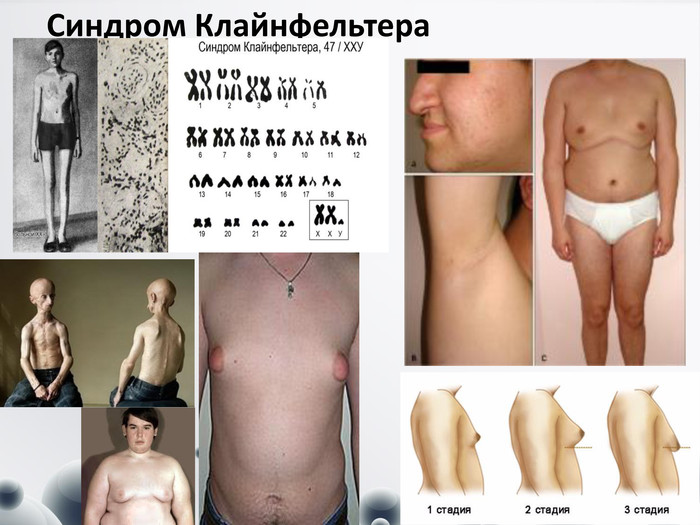
Синдром Клайнфельтера

Синдром Клайнфельтера Частота 1:400. Синдром виявляється лише в осіб чоловічої статі переважно при статевому дозріванні. Клінічні діагностичні ознаки:- високий зріст, - довгі кінцівки, - євнухоїдизм (широкий таз, вузькі плечі), - гінекомастія (збільшення молочних залоз), - відсутність сперматогенезу, - недорозвинення статевих залоз. - Тільця статевого хроматину виявляються в 80 % випадків. Інколи хворі з синдромом Клайнфельтера мають 48 і 49 хромосом (48, ХХXY; 49, ХХХХY). Чим більше Х-хромосом у каріотипі, тим вища ймовірність розвитку розумової відсталості. слабкий ріст волосся на обличчі, у пахвах і на лобкуяєсники зменшені у розмірах, відзначається статевий інфантилізм, схильність до ожеріння
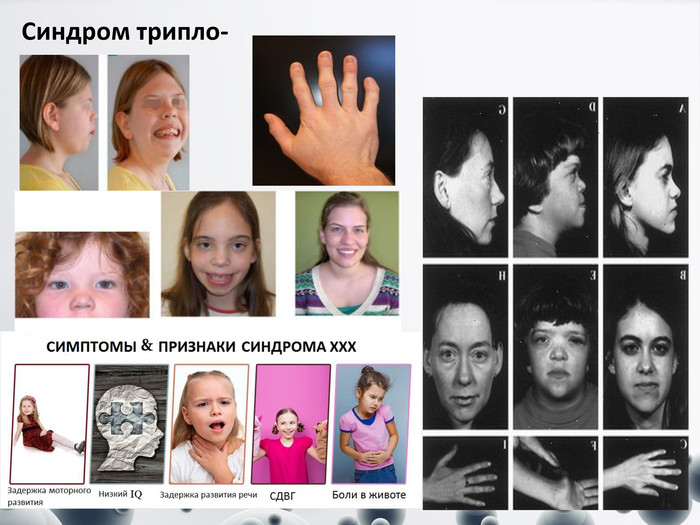
Синдром трипло-Х

Синдром трипло-Х Переважна більшість таких жінок мають нормальний фізичний та розумовий розвиток і виявляються випадково при обстеженні. Лише в деяких з них порушена репродуктивна функція. Більшість жінок мають нормальну плодючість, хоча зростає ризик мимовільних викиднів і хромосомних аберацій у нащадків, хромосомний набір – 47 хромосом з трьома Х-хромосомами (47 XXX) У хворих з трипло-Х спостерігаються:недорозвинені яєчники;гіпоплазована матка;безпліддя;нерегулярний менструальний цикл;знижене число примордіальних фолікулів;в них рано настає вторинна аменорея чи буває передчасний клімакс;хворі розумово відсталі, безплідні;діагноз грунтується на визначенні статевого хроматину, що дозволяє відрізнити хворих з аномальним комплексом Х-хромосом від хворих з первинною ендокринною патологією;кінцевий діагноз встановлюється при цитогенетичному дослідженні.
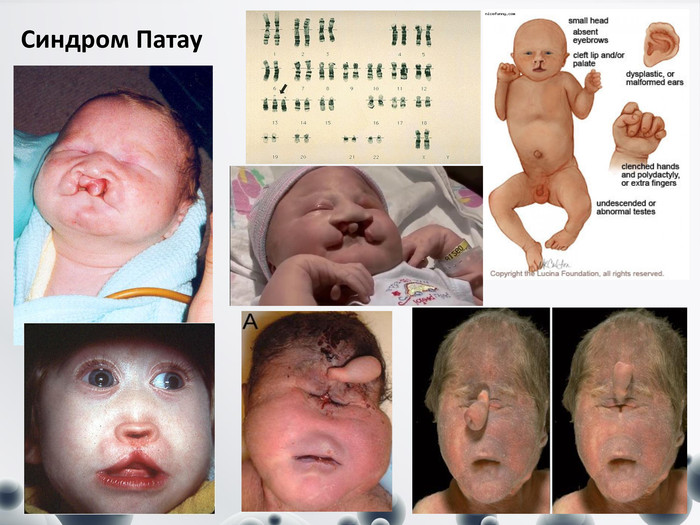
Синдром Патау

Синдром Патау (трисомія-13), серед різних трисомій трапляється найчастіше. Популяційна частота коливається в межах 1:7000-1:8000, у дівчаток спостерігається частіше, 1:4000. Хворі народженні мають нормальні розміри тіла та масу. Клінічні діагностичні ознаки: -щілини верхньої губи і піднебіння, - зменшений об’єм черепа, - перекошений, низький лоб, - мікрофтальмія, - анофтальмія (відсутність одного або обох очних яблук), - перенісся запале, - деформовані вушні раковини, полідактилія; - вроджені вади серця, інших внутрішніх органів. Більшість дітей вмирає в перші тижні або місяці. Вирішальним у діагностиці є цитогенетичне дослідження.
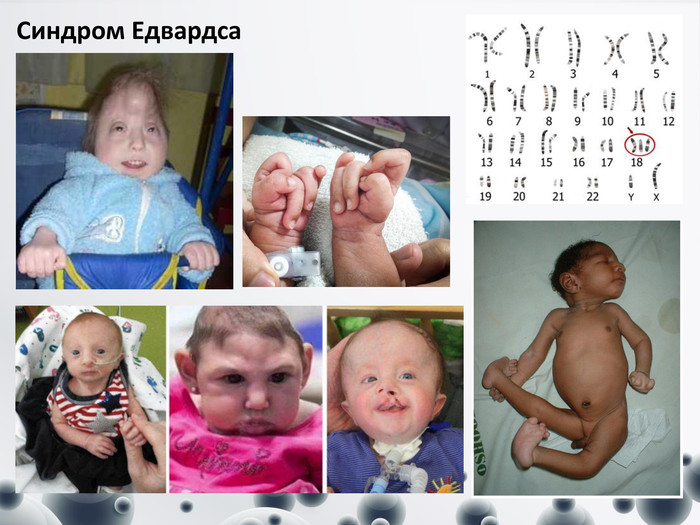
Синдром Едвардса

Синдром Едвардса Синдром Едвардса (трисомія-18). Частота 1:5000-1:7000. Співвідношення хлопчиків і дівчаток дорівнює 1:3. Причини переважання хворих дівчаток поки що невідомі. Клінічні діагностичні ознаки: - доліхоцефалічний череп, малі рот і нижня щелепа, - очні щілини вузькі, - вушні раковини деформовані, - аномальна стопа (“стопа-качалка”), - вроджені вади серця, скелетної системи, нирок, статевих органів. - Діти переважно вмирають до 2 місяців, 7% доживають лише до року. - Клінічний і навіть патологоанатомічний діагноз синдрому складні. Тому у всіх випадках показане цитогенетичне дослідження.
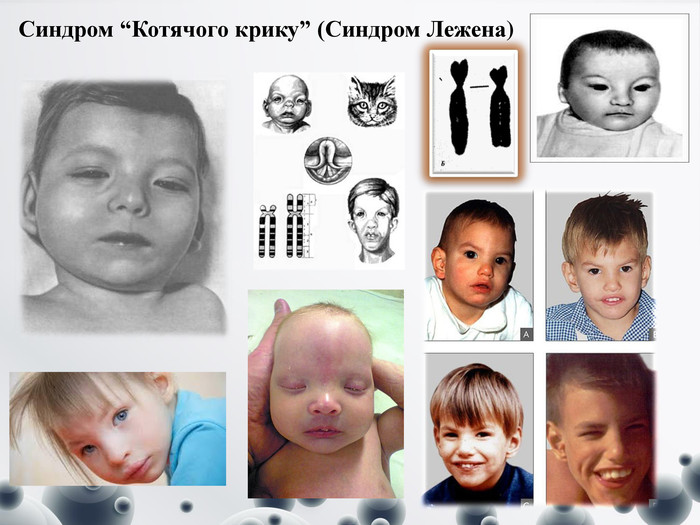
Синдром “Котячого крику” (Синдром Лежена)

Синдром “Котячого крику” Синдром Котячого крику виникає внаслідок втрати короткого плеча фрагмента 5-ї хромосоми. Плач хворих малюків нагадує м`явкання котів– обумовлена змінами гортані (звуження, м’якість хрящів, зменшеня надгортанника, незвична складчастість слизової оболонки), полягає в порушенні будови гортані, нявкаючий тембр голосу, розумова відсталість. Признак исчезает к концу первого года жизни. Частота синдрома примерно 1:45000 Клінічні прояви даного синдрому :- Практично у всіх хворих є зміни мозкового черепа та обличчя: - місяцеподібне обличчя, - мікроцефалія, - епікант, - антимонголоїдний розріз очей, - високе піднебіння, недорозвинення нижньої щелепи, плоска спинка носа. - Вушні раковини розмішені низько та деформовані- зустрічаються вроджені вади серця та інших внутрішніх органів, - опорно-рухового апарату (синдактилія стоп, косолапість). - м’язева гіпотонія, місяцеподібне обличчя з віком зникають майже повністю, а мікроцефалія є більш помітною, - прогресує розумова відсталість, - помітніше проявляється косоокість. - Більшість хворих помирає в перші роки, тільки 10 % досягають 10 річного віку.- низька вага тіла;- дефекти розвитку пальців;- клишоногість.
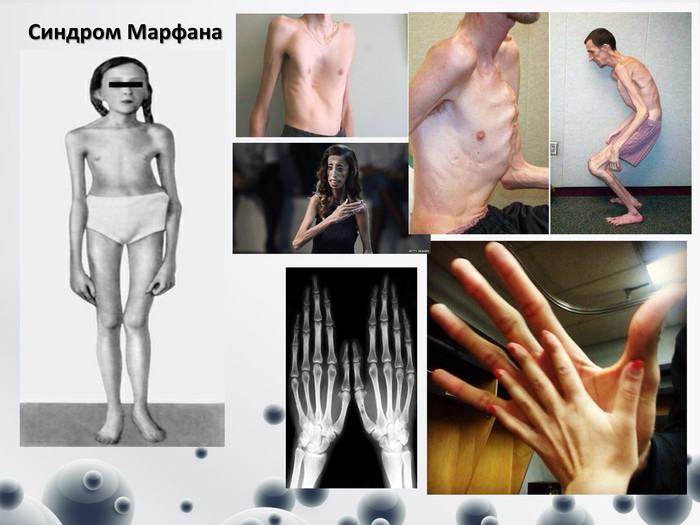
Синдром Марфана

Синдром Марфана(вроджена мезодермальна дистрофія) Вперше ця патологія була описана в 1896 році французьким педіатром А. Марфаном, який виявив характерну деформацію скелета у 5-річної дівчинки (довгі трубчасті кістки, павукоподібні пальці рук, високорослість). Хвороба Марфана виникає в результаті порушення синтезу фібриліну-1, який є основним структурним білком сполучної тканини. Цей синдром обумовлений мутацією гена, що кодує продукцію цього глікопротеїну. Синдром Марфана у дітей належить до рідкісних вроджених аномалій. Частота діагностованих випадків становить 1:10 000 – 1:20 000. Від хвороби Марфана частіше страждають хлопчики. Клінічні прояви даного синдрому:- ураженням скелета - подовження трубчастих кісток, доліхостеномелія, арахнодактилія, гіпермобільність суглобів, - очей (міопія, підвивих кришталика) - серцево-судинної системи (пролапс мітрального клапана, аневризма аорти).
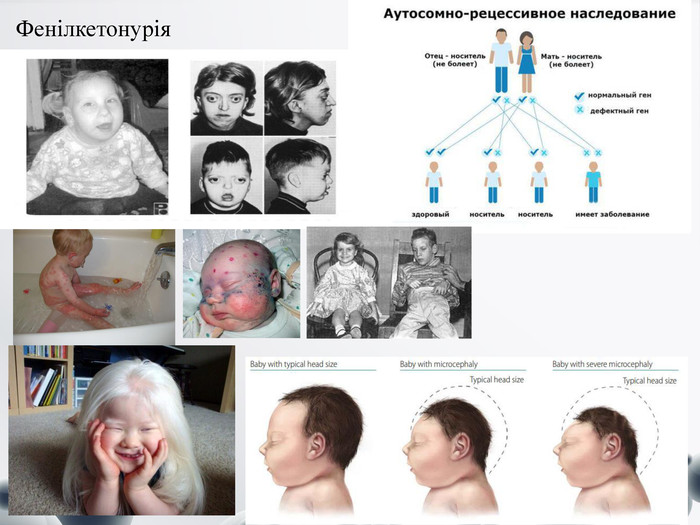
Фенілкетонурія

Фенілкетонурія Спадкова хвороба, яка зумовлена дефектом гена ферменту фенілаланінгідроксилази, що знаходиться на довгому плечі 12 хромосоми . Діти, народжені з фенілкетонурією, не здатні метаболізувати фенілаланін, який через це накопичується в крові. Така ненормальна висока кількість фенілаланіну перешкоджає нормальному розвитку мозку. За умови відсутності лікування, призводить до розумової відсталості. Спадкове захворювання, яке характеризується головним чином ураженням нервової системи. До об'єктивних ознак фенілкетонурії належать:- м'язова гіпертонія та гіперрефлексія (дитина сидить, підібгавши ноги під себе, під час ходьби ноги зігнуті в колінах і кульшових суглобах);- висипання на шкірі (папули, везикули), почервоніння шкіри;- інші шкірні симптоми: дермографізм, дерматит, екзема;- млявість реакції на навколишнє середовище;- порушення ковтання;- спонтанні судоми;- «мишачий» запах від шкіри та сечі;- зниження інтелекту.- Більшість дітей з даним захворюванням мають біле волосся та блакитні очі (ознаки альбінізму). Якщо не дотримуватися дієти, клінічні прояви ферментопатії наростають, і можуть призвести до олігофренії або ідіотії.
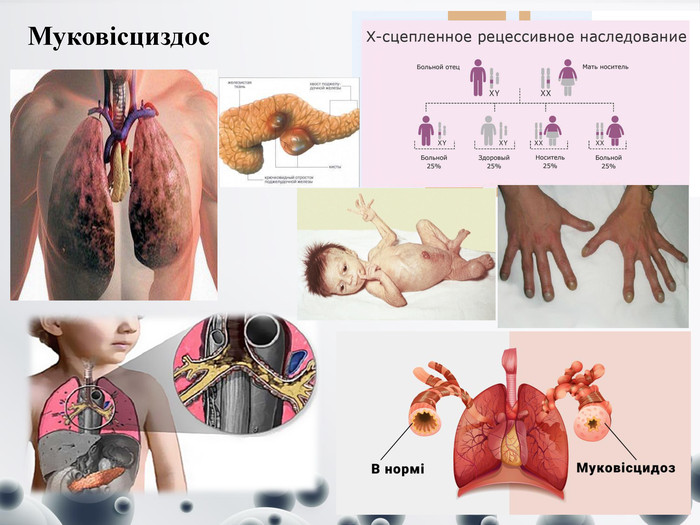
Муковісциздос

Муковісциздос (МВ, кістозний фіброз) (від лат. mucus-слиз, viscidus-липкий) — це спадкове аутосомно-рецесивне захворювання у дітей, що розвивається внаслідок продукції екзокринними залозами секрету підвищеної в'язкості та проявляється вторинними змінами у бронхолегеневій, травній та репродуктивній системах організму. Середня частота народження хворих дітей в європейських країнах складає 1:2500-3000, в Україні – в середньому 1:2300. До недавнього часу середня тривалість життя таких дітей становила до 5 років. Але впровадження в практику медико-генетичного консультування, пренатальної та постнатальної діагностики, сучасних методів лікування значно збільшили цей показник. Деякі пацієнти, хворі на МВ, доживають до 35-40 років. Симптоми:- Кашель (спочатку сухий, нападоподібний, такий, що нагадує епізоди коклюшу, потім – вологий, з відходженням в'язкого мокротиння). У старших дітей кашель найбільш виражений вранці та після фізичного навантаження. - дихання жорстке; за ателектазів і пневмонії – ослаблене; за масивних інфільтратів – бронхіальне; за численних бронхоектазів – амфоричне. Наявність постійних вологих дифузних хрипів, крепитуючих у разі загострення. Ускладнення – пневмоторакс, легенева кровотеча, деструкція легенів (булли, абсцеси та ін.).- інспіраторна задишка, цианоз, сухі свистячі хрипи, утворення «бочкоподібної» грудної клітки, поза ортопное, розвиток дихальної недостатності.- деформація пальців у вигляді «барабанних паличок» і нігтів у вигляді «годинникових скелець», периферичний цианоз, періостити в дистальних відділах довгих трубчастих кісток.- Зниження маси тіла при нормальному апетиті.
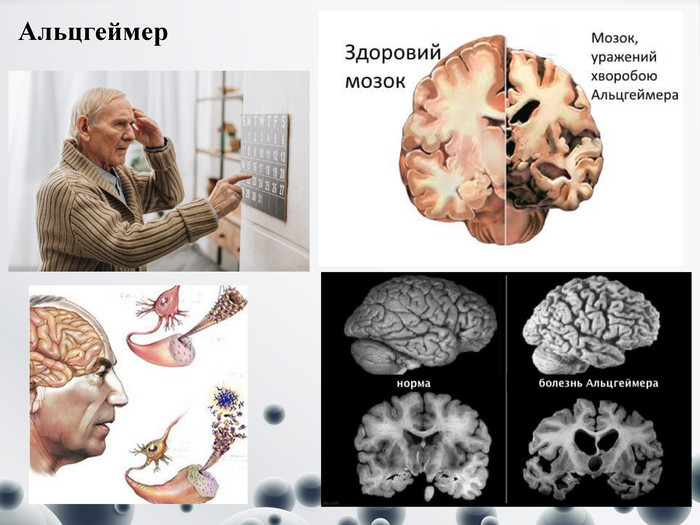
Альцгеймер

Альцгеймер Один з різновидів деменції, що уражає близько 6 % (одного з 16) людей віком понад 65 років. Названа на честь німецького психіатра і невролога Алоїза Альцгеймера. Про хворобу Альцгеймера зараз відомо, що вона виникає через аномальне накопичення певних білків у клітинах мозку і міжклітинному просторі. Один з цих білків — амілоїд, який відкладається у вигляді бляшок навколо нервових клітин. Другий — так звана аномальна білкова структура тау, або тау-білок, яку видно у вигляді клубків у самих нервових клітинах Симптоми:- присутня втрата місцезнаходження предметів у місці проживання;- забуваються імена друзів або потрібне слово у розмові;- людина може загубитися у знайомому місці або на знайомому маршруті;- забуваються зустрічі і знаменні дати.- мовлення. Повторення одних і тих же фраз, фактів, підвищена увага до розмови через побоювання не вловити сенс;- з'являються проблеми з оцінкою відстані до предметів, проблеми в розпізнанні форми предметів;- підніматися або спускатися сходами або припаркувати машину також стає набагато важче;- концентрація, планування або організація – таким людям складно приймати рішення, вирішувати проблеми або виконувати послідовність завдань (наприклад, готувати їжу);- порушення орієнтації на місцевості або у часі.
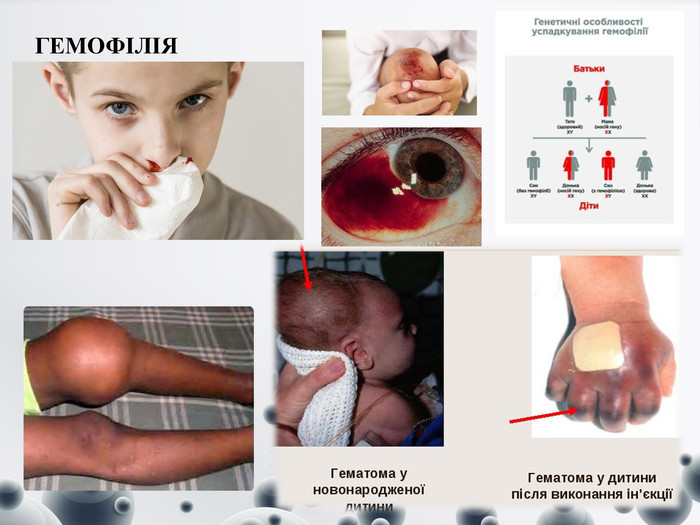
ГЕМОФІЛІЯ

ГЕМОФІЛІЯ Це рідкісна вроджена патологія, обумовлена порушенням нормального згортання крові. У дітей з цим захворюванням часто виникають спонтанні й індуковані кровотечі, які погано зупиняються і можуть спровокувати розвиток тяжких ускладнень (крововилив у головний мозок, черевну порожнину, суглоби). Поширеність цієї патології становить 1:5000–10000. Хворіють виключно хлопчики, а дівчатка можуть бути тільки носіями дефектного гена. Загальні: - слабкість, ортостаз (гіпотензія при вертикальному положенні тіла), тахікардія, тахіпное.- Скелетно-м'язові: кольки, біль, тріск, жар, скутість у суглобах.- ЦНС: головний біль, ригідність потиличних м'язів, блювання, млявість, дратівливість та синдром ураження спинного мозку.- ШКТ: гематемезис (кривава блювота), мелена (дьогтьоподібний кал), виділення яскравої червоної крові з прямої кишки, біль у животі.- Урогенітальні: гематурія, ниркова коліка.- Інші: носові кровотечі, кровотечі у слизових оболонках ротової порожнини, кровохаркання, задишки (гематоми призводять до обструкції дихальних шляхів), надмірна кровотеча після проведення рутинних стоматологічних процедур тощо.
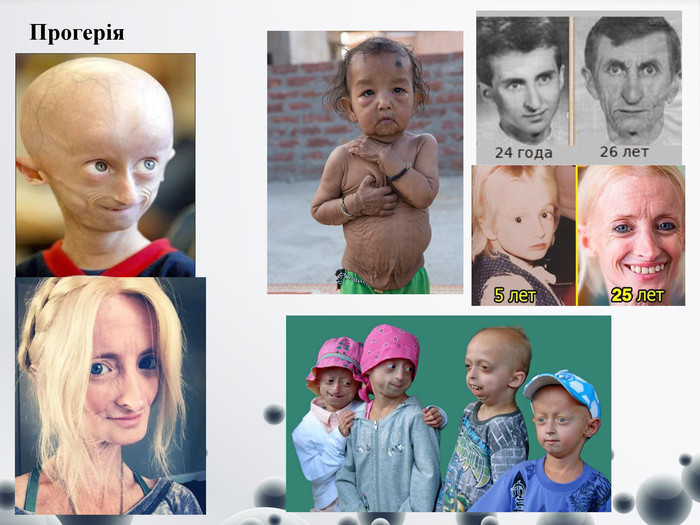
Прогерія

Прогерія Прогерия (синдром Хатчінсона-Гілфорда) – рідкісна патологія, спричинена мутацією гена, відповідального за синтез білка. При такій патології з’являються зміни шкіри і внутрішніх органів, що викликані передчасним старінням. Дитяча прогерия, симптоми якої з’являються з віку 2 років, викликає передчасне старіння: хворі доживають в середньому до 13 років і вмирають від атеросклерозу і пов’язаних з ним захворювань, інсульту, інфаркту міокарда. Незважаючи на генетичний характер хвороби, у спадок не передається. Доросла форма – синдром Вернера – генетична патологія, передається у спадок, починається після 18 років, характеризується раннім старінням, розвитком хвороб літнього віку: атеросклерозу, остеопорозу, катаракти. Призводить до летального результату. Синдром Хатчінсона-Гілфорда – це наслідок мутації, зміни структури гена, яке відбувається спонтанно або під впливом зовнішніх факторів. Носієм спадковості людини є молекула ДНК. Дитяча прогерия має наступні симптоми:- маленький зріст;- відсутність підшкірної клітковини;-розширена відень під шкірою;-непропорційно великий череп;-відсутність волосся на голові;-поганий фізичний розвиток;-великі очі;-дефекти зубів;-«кілеподібна груди»;-високий голос.-Випадки прогерии у дорослих, тобто синдром Вернера, характеризуються наступними станами:-рання сивина і облисіння;-поява старечих зморщок у молодому віці;-пігментація, сухість шкіри;-фіброзні ущільнення в підшкірній клітковині;-голос стає глухим.
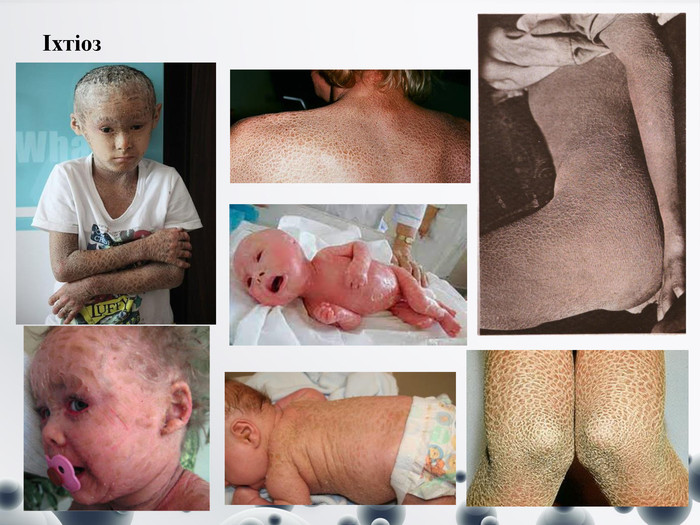
Іхтіоз

Іхтіоз Це досить велика група спадкових захворювань шкіри, що проявляються в порушенні процесів зроговіння. Ці захворювання характеризуються появою на шкірі утворень, що нагадують луску риби. Іхтіоз у медичній практиці має безліч синонімів, наприклад, «шкіра алігатора», «кератома», «сауріаз» та ін. Це захворювання носить спадковий характер, основною причиною його появи є генна мутація. Основною причиною його виникнення вважається генна мутація. До чинників, які можуть спровокувати дебют хвороби, належать:-авітаміноз, особливо дефіцит вітаміну А;-імунодефіцитні стани;-ендокринні порушення;-онкологічні хвороби. Основним його симптомом -є поява на шкірі утворень, дуже схожих на «риб'ячі лусочки», які можуть бути:-невеликого розміру – найчастіше, покривають всю шкіру;-великого розміру, сірувато-коричневого кольору;-лусочки, які розташовуються за типом мозаїки, сірувато-прозорого кольору. Також у хворого можуть виникнути такі скарги:-сухість і шорсткість шкірних покривів;-дрібнопластинчасте лущення шкіри;-відчуття стягування шкіри;-посилення шкірного малюнка на долонях і підошвах;-помірне свербіння шкіри;-ламкість, розшарування, зміна кольору та форми нігтів;-витончення і випадіння волосся.

ДАЛЬТОНІЗМ

ДАЛЬТОНІЗМ Спадковий дальтонізм зустрічається частіше, вражає обидва ока і не погіршується з часом. Цей варіант дальтонізму в різній ступені вираженості присутній у 8% чоловіків і 0,4% жінок. Спадковий дальтонізм пов'язаний з X-хромосомою та практично завжди передається від матері-носія гена до сина. Дальтонізм може бути частковим (пов'язаним з окремими кольорами) або повним (пов'язаним з усіма кольорами). Повний дальтонізм зустрічається дуже рідко і, як правило, в поєднанні з іншими серйозними вродженими дефектами очей. При частковому дальтонізмі зустрічаються:-Проблеми з розрізненням червоного та зеленого кольорів (найбільш часто).-Проблеми з розрізненням синього та зеленого кольорів (менше часто). -Симптоми більш серйозних вроджених (рідко - набутих) форм дальтонізму можуть включати:-Всі предмети виглядають пофарбованими в різні відтінки сірого.-Низька гострота зору.-Ністагм.

Альбінізм

Альбінізм Альбіні́зм (від лат. albus — білий) — уроджена відсутність пігменту шкіри, волосяного покриву, пір'я, райдужки ока у тварин. Наприклад, у бджіл альбінізм характеризується відсутністю пігмента в очах (білоокі трутні). Вважається, що причиною захворювання є відсутність (або блокада) ферменту тирозинази, необхідної для нормального синтезу меланіну — особливої речовини, від якої залежить забарвлення тканин. Отже, білий колір альбіносів — не забарвлення, а його відсутність. Альбінізм може спостерігатися тільки у тих осіб, обоє батьків яких мають відповідний ген, при цьому ймовірність народження дитини-альбіноса при кожній вагітності складає 25 %. Види альбінізму Виділяють тотальний, неповний і частковий альбінізм
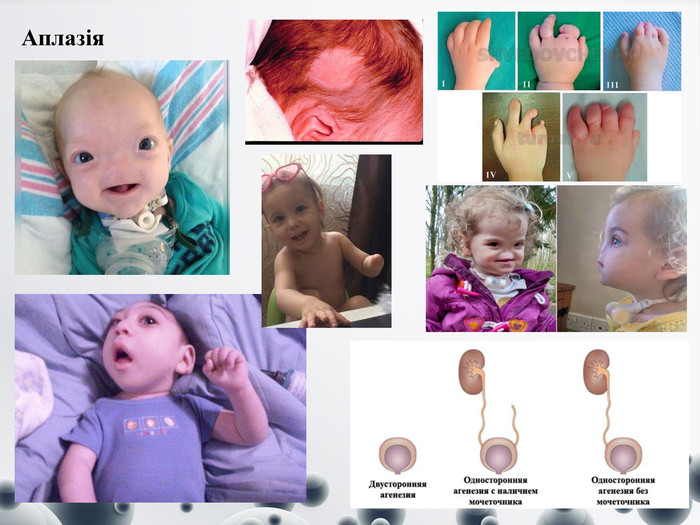
Аплазія

Аплазіяодин із проявів дефектів розвитку, що характеризується відсутністю всього органа, його частини, ділянки тканини, частини тіла або всього зародка.


про публікацію авторської розробки
Додати розробку