Презентація "Спадкові захворювання людини. Медико – генетичне консультування".


Спадкові захворювання людини. Медико – генетичне консультування. Волошина Олена Іванівна Вчитель біології вищої категоріїХарківської ЗОШ № 125
Уявіть таку картину: молодята мріють про дитину і відбувається магія, зароджується новий організм. Спочатку це лише клітина, у хромосомах якої записана вся історія роду жінки та чоловіка. Якою буде ця дитинка, який у неї характер, здібності, талант?І раптом… сумна звістка, діагноз дитини – спадкова хвороба. Батьки у відчаї, почувають себе винними перед дитиною. А може ця дитина не хвора, а «особлива»? Хто ж вони – «особливі» діти? В арсеналі сучасної медицини існують методи діагностики, профілактики і лікування багатьох спадкових патологій. Що ж таке аномалії у розвитку дитини, вроджені вади, що їх спричиняє. Чи можливо запобігти народженню хворої дитини, наскільки сучасна наука дає можливість попередити це страшне лихо, ми дізнаємось сьогодні на уроці.
Спадкові хвороби людини- захворювання, викликані хромосомними і генними дефектами. Основою спадкових захворювань є генні, хромосомні і мітохондріальні порушення спадкової інформації. Не варто плутати спадкові та вроджені захворювання. Вроджені захворювання обумовлені не тільки спадковими, але і зовнішніми чинниками, наприклад негативним впливом на ембріон хімічних речовин, ліків або опромінення. Спадкові захворювання людини можуть бути виявлені відразу після народження, а можуть проявитися через тривалий час. Близько 10% всіх захворювань людини обумовлено патологічними генами або генами, які відповідають за схильність до хвороби.
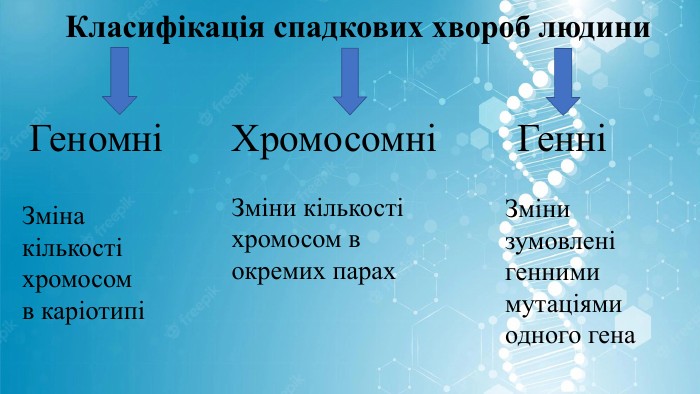
Класифікація спадкових хвороб людини. ГеномніХромосомні ГенніЗміни зумовлені генними мутаціями одного гена Зміни кількості хромосом в окремих парах. Зміна кількості хромосом в каріотипі

Геномні та хромосомні захворювання. Види геномних мутацій: Причиною геномних хвороб, є нерозходження хромосом при поділі клітини. Поліплоїдія Гетероплоїдію (анеуплоїдію)Збільшення або зменшення кількості хромосом на 1, 2 або більше. Види гетероплоїдії:а) Моносомія — відсутність однієї хромосоми (2n-1).б) Нулісомія — відсутність однієї пари хромосом (2n-2), летальна мутація в більшості випадківв) Трисомія — одна зайва хромосома (2n+1).г) Тетрасомія — дві зайві хромосоми (2n+2).д) Пентасомія — три зайві хромосоми (2n+3). Збільшення кількості хромосом на величину, кратну гаплоїдному набору (3n, 4n, ...). У людини описана триплоїдія (3n=69 хромосом) і тетраплоїдія (4n=92 хромосоми).

Причиною дефіциту чи надлишку хромосомного матеріалу в клітині можуть бути хромосомні перебудови, що виникають при розривах хромосом та їх з’єднанні в іншому порядку. Відновлення розривів може привести до переміщення ділянок хромосом, їх подвоєння чи повороту всередині хромосоми. Можуть також виникати обміни ділянками між негомологічними хромосомами. Хромосомні перебудови в людини призводять до множинних пороків розвитку. Внаслідок цього виникають так звані хромосомні хвороби. Деякі з них передаються потомству. На відміну від анеуплоїдій, при хромосомних перебудовах кількість хромосом не змінюється.
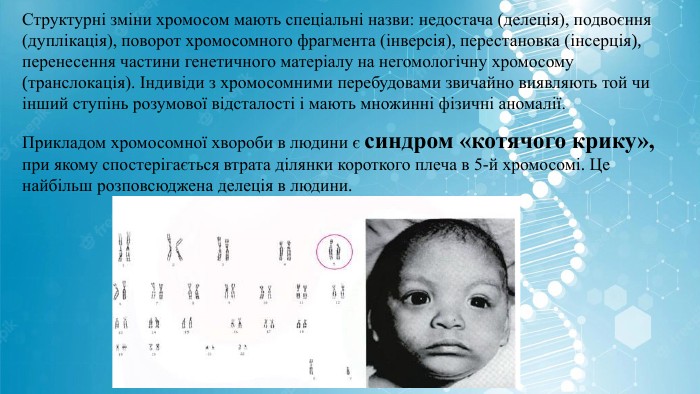
Структурні зміни хромосом мають спеціальні назви: недостача (делеція), подвоєння (дуплікація), поворот хромосомного фрагмента (інверсія), перестановка (інсерція), перенесення частини генетичного матеріалу на негомологічну хромосому (транслокація). Індивіди з хромосомними перебудовами звичайно виявляють той чи інший ступінь розумової відсталості і мають множинні фізичні аномалії. Прикладом хромосомної хвороби в людини є синдром «котячого крику», при якому спостерігається втрата ділянки короткого плеча в 5-й хромосомі. Це найбільш розповсюджена делеція в людини.

Більшість ембріонів із хромосомними аномаліями нежиттєздатні і мимовільно абортуються. Усі хромосомні аномалії пов’язані з порушенням інтелекту і поведінки. Хромосомні хвороби не виліковуються, тому єдиний спосіб їхньої профілактики — запобігання народженню хворих дітей. Більшість хромосомних хвороб не є спадковими в тому розумінні, що вони не пере-даються від батьків до дітей, оскільки хворі, як правило, помирають у дитинстві, а якщо доживають до репродуктивного віку, то звичайно виявляються бесплідними. У зв’язку з цим хромосомні хвороби являють собою нові мутації, що виникають у статевих клітинах батьків. Більшість ознак і хвороб людини залежать не від одного чи двох генів, а від цілого комплексу генів і в той же час від умов навколишнього середовища.

Вважається, що при клітинному розподілі мітохондрії випадково розподіляються між дочірні-ми клітинами. Для мітохондріальних хвороб характерна різна експресивність (сила прояву), оскільки фенотипічний прояв патологічного гена залежить від співвідношення нормальних і мутантних мітохондрій. Серед мітохондріальних хвороб найкраще вивчений синдром Лебера. Мітохондріальні хвороби успадковуються тільки по материнській лінії.

Найпоширенішою аутосомною анеуплоїдією є синдром Дауна. Причиною синдрому Дауна є трисомія за 21-ю хромосомою. Діти із синдромом Дауна мають множинні вади розвитку і на-роджуються з частотою 1:700. У молодих матерів імовірність народження дитини з цим синд-ромом становить 1:2000, у жінок старше 40 років — 1:22. Характерні симптоми хвороби – це специфічні риси обличчя: типове плоске перенісся, велика кругла голова, маленький зріст, гіпотонія м'язів, розумова відсталість, короткі кінцівки та вузькі розкосі очі. Такі діти повільніше розвиваються, мають низький імунітет. Чоловіки безплідні, жінки можуть народити дитину, але ймовірність народити дитину з таким же діагнозом висока.
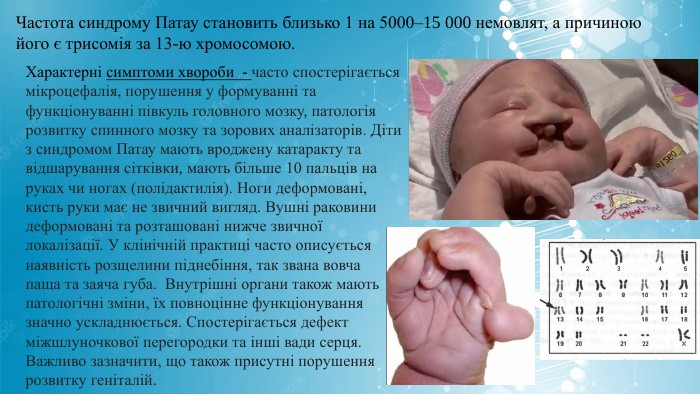
Частота синдрому Патау становить близько 1 на 5000–15 000 немовлят, а причиною його є трисомія за 13-ю хромосомою. Характерні симптоми хвороби - часто спостерігається мікроцефалія, порушення у формуванні та функціонуванні півкуль головного мозку, патологія розвитку спинного мозку та зорових аналізаторів. Діти з синдромом Патау мають вроджену катаракту та відшарування сітківки, мають більше 10 пальців на руках чи ногах (полідактилія). Ноги деформовані, кисть руки має не звичний вигляд. Вушні раковини деформовані та розташовані нижче звичної локалізації. У клінічній практиці часто описується наявність розщелини піднебіння, так звана вовча паща та заяча губа. Внутрішні органи також мають патологічні зміни, їх повноцінне функціонування значно ускладнюється. Спостерігається дефект міжшлуночкової перегородки та інші вади серця. Важливо зазначити, що також присутні порушення розвитку геніталій.

Трисомія за 18-ю хромосомою призводить до синдрому Едвардса, що має летальний наслідок. Діти із синдромом Едвардса народжуються з частотою приблизно в 1 на 4000–8000. Характерні симптоми хвороби - череп здавлений з боків, з низьким чолом і широкою виступаючою потилицею; іноді зустрічається мікроцефалія або гідроцефалія. Надочні валки згладжені, очні щілини вузькі, спостерігають очну патологію, катаракту. Перенісся втиснене, але спинка носа тонка, виступає, вушні раковини розташовані дуже низько, часто відсутні мочка. Рот маленький, трикутної форми з короткою верхньою губою; піднебіння високе, іноді з щілиною, шия коротка, часто з крилоподібною складкою.

В основі синдрому Клайнфельтера лежить каріотип 47,ХХY. У середньому частота синд-рому Клайнфельтера в популяції досить висока — 1 хлопчик на 1000 немовлят має це захворю-вання. Зайва Y-хромосома в чоловіків зустрічається з частотою приблизно 1:1000. Часто прояви не мають значного розвитку, багато людей не підозрюють, що мають таку хворобу. Іноді прояви помітніші, часто є м'язова слабкість, високий зріст, погана координація, рідке оволосіння шкіри, ожиріння, остеопороз, малі статеві органи, збільшення молочних залоз, зменшення статевого потягу.

Хворооба Гаантінгтона (відома також як Хорея Гаантінгтона)— генетичне захворювання нервової системи, що уражає людину у віці 35-50 років внаслідок змін у гені IT-15, що знаходиться на 4-ій хромосомі, яке призводить до атрофії стріатума, а на пізній стадії — до атрофії кори головного мозку. Розвиток хвороби швидкий, але поступовий. Симптоми малої хореї у дітей:щебет губами під час розмови, часті і глибокі зітхання, постійне шмигання носом, спонтанне випинання язика. Мімічні мимовільні рухи на обличчі поступово стають інтенсивнішими, до них приєднуються пританцьовування, недоречні кивання головою, розмахування руками, хитання корпусом з боку в бік.

Генні хвороби — спадкові патології, які спричинені мутацією одного гена і передаються наступним поколінням за законами Менделя. Синдром Марфана Холта — Орама (синдром "рука—серце")Муковісцидоз Фенілкетонурія. Дальтоніз
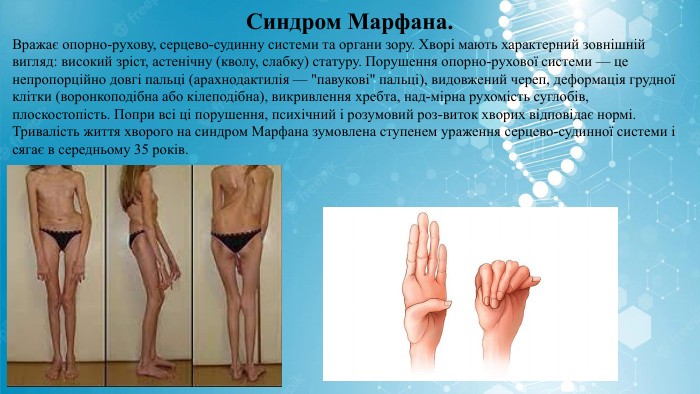
Синдром Марфана. Вражає опорно-рухову, серцево-судинну системи та органи зору. Хворі мають характерний зовнішній вигляд: високий зріст, астенічну (кволу, слабку) статуру. Порушення опорно-рухової системи — це непропорційно довгі пальці (арахнодактилія — "павукові" пальці), видовжений череп, деформація грудної клітки (воронкоподібна або кілеподібна), викривлення хребта, над-мірна рухомість суглобів, плоскостопість. Попри всі ці порушення, психічний і розумовий роз-виток хворих відповідає нормі. Тривалість життя хворого на синдром Марфана зумовлена ступенем ураження серцево-судинної системи і сягає в середньому 35 років.
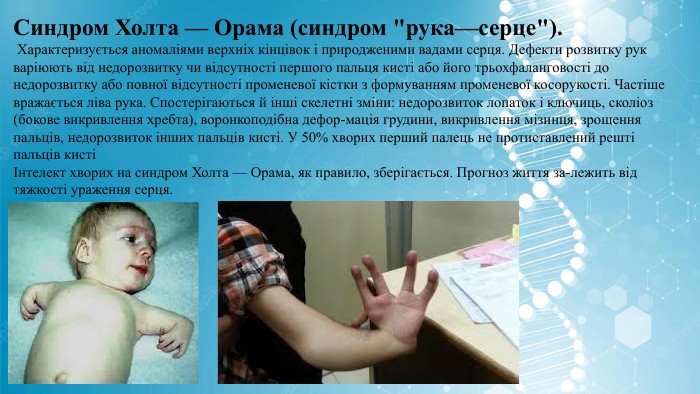
Синдром Холта — Орама (синдром "рука—серце"). Характеризується аномаліями верхніх кінцівок і природженими вадами серця. Дефекти розвитку рук варіюють від недорозвитку чи відсутності першого пальця кисті або його трьохфаланговості до недорозвитку або повної відсутності променевої кістки з формуванням променевої косорукості. Частіше вражається ліва рука. Спостерігаються й інші скелетні зміни: недорозвиток лопаток і ключиць, сколіоз (бокове викривлення хребта), воронкоподібна дефор-мація грудини, викривлення мізинця, зрощення пальців, недорозвиток інших пальців кисті. У 50% хворих перший палець не протиставлений решті пальців кисті Інтелект хворих на синдром Холта — Орама, як правило, зберігається. Прогноз життя за-лежить від тяжкості ураження серця.
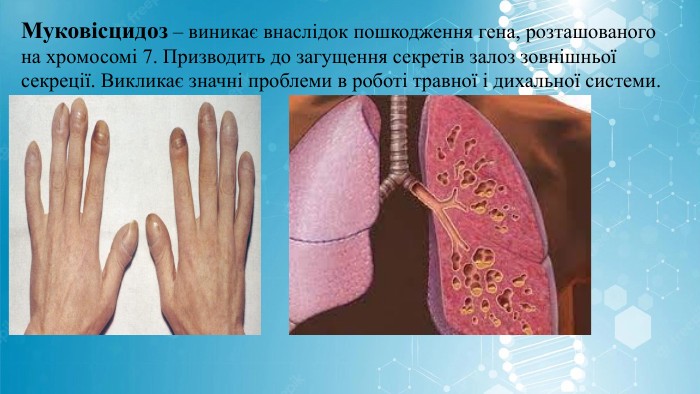
Муковісцидоз – виникає внаслідок пошкодження гена, розташованого на хромосомі 7. Призводить до загущення секретів залоз зовнішньої секреції. Викликає значні проблеми в роботі травної і дихальної системи.
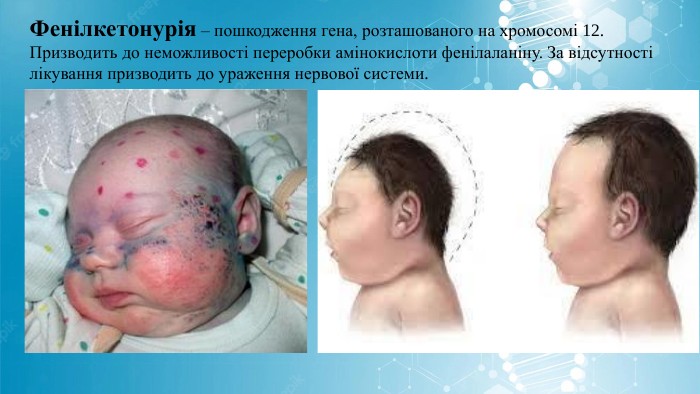
Фенілкетонурія – пошкодження гена, розташованого на хромосомі 12. Призводить до неможливості переробки амінокислоти фенілаланіну. За відсутності лікування призводить до ураження нервової системи.

Дальтонізм – пошкодження одного з трьох генів, що відповідають за синтез білка опсину, який сприймає відповідний колір. Ген синього опсину розташований на хромосомі 7, а гени червоного і синього на Х-хромосомі. Призводить до втрати можливості сприймати відповідний колір.
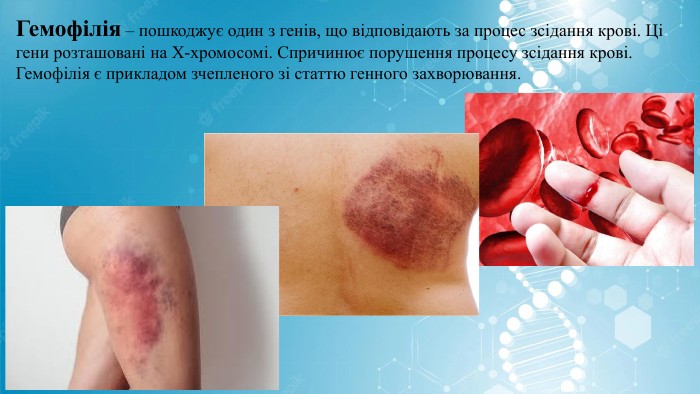
Гемофілія – пошкоджує один з генів, що відповідають за процес зсідання крові. Ці гени розташовані на Х-хромосомі. Спричинює порушення процесу зсідання крові. Гемофілія є прикладом зчепленого зі статтю генного захворювання.
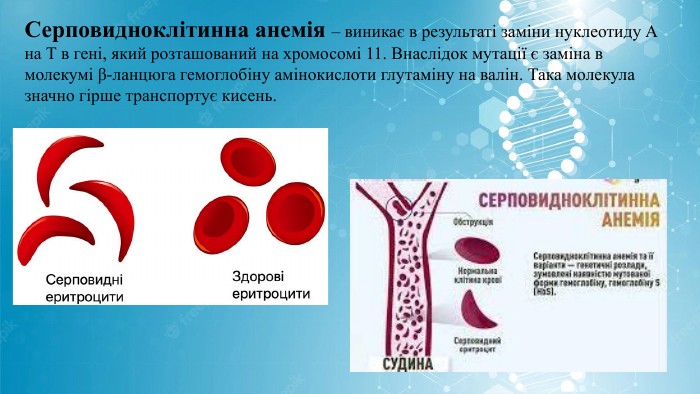
Серповидноклітинна анемія – виникає в результаті заміни нуклеотиду А на Т в гені, який розташований на хромосомі 11. Внаслідок мутації є заміна в молекумі β-ланцюга гемоглобіну амінокислоти глутаміну на валін. Така молекула значно гірше транспортує кисень.

Медико–генетичне консультування – це спеціальний вид медичної допомоги населенню, направлений на профілактику спадкових хвороб. Медико–генетичні консультації допомагають людям у прогнозуванні шлюбу, потомства, консультують вагітних жінок, допомагають у вирішенні питань спадковості, усиновлення дитини, проводять точну діагностику спадкових захворювань. Під час консультування визначається прогноз народження дитини зі спадковою патологією,пояснюється вірогідність цієї події і надається допомога у прийнятті рішення про народження.

Етапи медико-генетичного консультування
Генетичне консультування Активне Пасивне. При пасивній лікар-генетик дає поради особам, які самостійно звернулись до консультації. Роль лікаря у відборі осіб для консультування пасивна. Зарубіжні країни обмежуються лише цією формою консультації, що обумовлено комерційним характером медицини. Велика кількість осіб, яким показане МГК, не входить в коло обстежених. Активна форма МГК відповідає профілактичному напрямку медицини нашої країни. Лікувально-профілактичні заклади виявляють осіб, яким необхідне консультування, і направляють їх в МГК. Об’єктом активної консультації повинні бути насамперед здорові родичі хворих зі спадковою патологією. При цьому вирішується два питання: прогноз для їхнього потомства і виявлення схильності до сімейної патології у всіх членів родини.

Якщо консультування проводять до настання вагітності, то після з'ясування кількісної оцінки ризику спадкової та вродженої патології у майбутніх дітей родина одержує такі рекомендації:— оптимальні заходи підготовки до вагітності;— індивідуальне спостереження за майбутньою матір'ю в період вагітності;— можливі способи підтвердження чи спростування конкретного спадкового захворювання плода в першому-другому триместрах вагітності. Якщо консультування проводять уже під час вагітності, то йдеться лише про рекомендації стосовно спостереження та пренатальної діагностики. Однак оптимальним часом для звертання до медико-генетичної консультації є період, коли родина ще тільки планує народження первістка. У разі виявлення захворювання батьки мають змогу прийняти рішення про припинення чи продовження вагітності. Останнім часом з'явилася можливість за допомогою технології запліднення "у пробірці" поставити діагноз ембріону ще до перенесення його в порожнину матки.
Критерієм ефективності медико-генетичного консультування в широкому розумінні є зменшення частоти патологічних алелей, а окремої консультації — зміна поведінки подружніх пар, що звертаються з питань дітородіння. За широкого впровадження медико-генетичного консультування можна досягнути деякого зниження частоти спадкових хвороб, а також смертності (особливо дитячої) у популяції.



про публікацію авторської розробки
Додати розробку