Урок "Спадкові захворювання"

Спадкові хвороби людини
Цілі уроку:
освітня: познайомити учнів з особливостями генетики людини та методами, які використовуються при її вивченні, розглянути спадкові хвороби людини та способами їх профілактики;
розвивальна: розвивати вміння виявляти закономірності спадковості та мінливості живих організмів, вміння логічного мислення
виховна: виховувати розуміння єдності всіх біологічних процесів у живих організмів та їхньої важливості для існування життя
Обладнання: підручник, фотографії людей із спадковими вадами,
Базові поняття і терміни: генетика людини, методи генетичних досліджень, спадкові захворювання
Тип уроку: засвоєння нових знань
Ключові компетентності: володіння державною мовою, основні компетентності у природничих науках і технологіях, екологічна грамотність і здорове життя, уміння вчитися впродовж життя.
Хід уроку
- Організація навчальної діяльності
- Актуалізація опорних знань
- Що таке мутації?
- Які види мутацій ви знаєте?
- Яке значення має мутаційна мінливість для живих організмів?
- Які фактори можуть бути мутагенними?
- Мотивація навчальної діяльності
Цікава статистика: 40% випадків самовільного переривання вагітності пов’язані з хромосомними аномаліями. Хромосомні захворювання спостерігаються у 1% новонароджених. Вони є причиною 45-50% множинних уроджених вад розвитку, 36% випадків розумової відсталості, 50% безпліддя у жінок та 10% безпліддя у чоловіків. Генних захворювань нараховують понад 3500. Близько 5% дітей народжуються з генетичними дефектами. Генні хвороби складають 8% всіх вад розвитку людини.
Які ж спадкові захворювання зустрічаються найчастіше.?
Про це ми поговоримо сьогодні на уроці
- Вивчення нового матеріалу
Генетика людини та спадкові захворювання
Явища спадковості та мінливості у популяціях людей, особливості успадкування нормальних і патологічних ознак, залежність захворювання відгенетичної схильності й факторів середовища вивчає генетика людини. Ця наука об´єднує кілька напрямів дослідження спадковості людини.
Медична генетика зосереджена на дослідженнях спадкових захворювань і порівнянні успадкування й реалізації нормальних і патологічних ознак та залежності захворювань від генетичних факторів.
Антропогенетика сформувалась на межі генетики людини, біології людини та антропології. Цей напрям досліджує процеси, які відбуваються у сучасних та проходили у давніх популяціях людини, залучаючи ряд інших методів дослідження.
Загалом дослідження генетики людини є досить складним, оскільки у ній не можливе експериментальне схрещування, повільна зміна поколінь, малій кількості нащадків, а ще складність каріотипу, велика кількість груп зчеплення.
Як і в інших живих організмів, генетичні порушення в організмі людини можуть відбуватися на різних рівнях організації спадкового матеріалу. А отже, трапляються геномні, хромосомні та генні захворювання
Розглянемо окремо кожен тип захворювань.
Геномні та хромосомні захворювання
Захворювання спричинені геномними та хромосомними порушеннями зазвичай дуже тажкі й рідко бувають сумісними з життям.
Причиною геномних хвороб, або анеуплоїдій, є нерозходження хромосом при поділі клітини. У нормі при мейозі пари гомологічних хромосом розходяться, й у кожному сперматозоїді чи яйцеклітині міститься тільки по одній хромосомі (гомологу) кожної пари. Нерозходження може відбуватися як при першому, так і при другому мейотичному поділі. Результатом нерозходження є гамета, що містить дві копії хромосоми чи не містить жодної. Кожна з 22 пар аутосом може мати додаткову чи відсутню хромосому, крім цього, статеві хромосоми можуть виявитися в анеуплоїдних комбінаціях (Y0, Х0, ХХХ, ХХY і ХYY). Однак лише деякі хромосомні патології зустрічаються у немовлят, а в основному в таких випадках розвиток припиняється ще в ембріональний період. Найпоширенішою аутосомною анеуплоїдією є синдром Дауна. Причиною синдрому Дауна є трисомія за 21-ю хромосомою. Діти із синдромом Дауна мають множинні вади розвитку і народжуються з частотою 1:700. У молодих матерів імовірність народження дитини з цим синдромом становить 1:2000, у жінок старше 40 років — 1:22.
Частота синдрому Патау становить близько 1 на 5000–15 000 немовлят, а причиною його є трисомія за 13-ю хромосомою. Трисомія за 18-ю хромосомою призводить до синдрому Едвардса, що має летальний наслідок.
Діти із синдромом Едвардса народжуються з частотою приблизно в 1 на 4000–8000. Частота синдрому Шерешевського - Тернера серед новонароджених дівчаток 1:2000. Причина цього захворювання — відсутність другої Х-хромосоми. Триплоїдія за Х-хромосомою, або синдром трипло-Х, зустрічається з частотою приблизно 1 на 1000–2000 жінок. Більшість жінок із синдромом трипло-Х фертильні.
В основі синдрому Клайнфельтера лежить каріотип 47,ХХY. У середньому частота синдрому Клайнфельтера в популяції досить висока — 1 хлопчик на 1000 немовлят має це захворювання. Зайва Y-хромосома в чоловіків зустрічається з частотою приблизно 1:1000. Раніше вважали, що чоловіки із зайвою Х-хромосомою агресивні, однак на сьогодні ця гіпотеза не підтвердилася.
Причиною дефіциту чи надлишку хромосомного матеріалу в клітині можуть бути хромосомні перебудови (аберації), що виникають при розривах хромосом та їх з’єднанні в іншому порядку. Відновлення розривів може привести до переміщення ділянок хромосом, їх подвоєння чи повороту всередині хромосоми. Можуть також виникати обміни ділянками між негомологічними хромосомами. Хромосомні перебудови в людини призводять до множинних пороків розвитку. Внаслідок цього виникають так звані хромосомні хвороби. Деякі з них передаються потомству. На відміну від анеуплоїдій, при хромосомних перебудовах кількість хромосом не змінюється.
Структурні зміни хромосом мають спеціальні назви: недостача (делеція), подвоєння (дуплікація), поворот хромосомного фрагмента (інверсія), перестановка (інсерція), перенесення частини генетичного матеріалу на негомологічну хромосому (транслокація). Індивіди з хромосомними перебудовами звичайно виявляють той чи інший ступінь розумової відсталості і мають множинні фізичні аномалії.
Прикладом хромосомної хвороби в людини є синдром «котячого крику», при якому спостерігається втрата ділянки короткого плеча в 5-й хромосомі. Це найбільш розповсюджена делеція в людини.
Прикладом багаторазової дуплікації (більше 36) одного з кодонів (CAG) гена ІТ – 15, розташованого на хромосомі 4 розвивається хорея Гентінгтона. Хвороба розвивається у віці 35-50 років і стає причиною атрофії клітин деяких відділів та кори головного мозку.
Більшість ембріонів із хромосомними аномаліями нежиттєздатні і мимовільно абортуються. Усі хромосомні аномалії пов’язані з порушенням інтелекту і поведінки. Хромосомні хвороби не виліковуються, тому єдиний спосіб їхньої профілактики — запобігання народженню хворих дітей. Більшість хромосомних хвороб не є спадковими в тому розумінні, що вони не передаються від батьків до дітей, оскільки хворі, як правило, помирають у дитинстві, а якщо доживають до репродуктивного віку, то звичайно виявляються бесплідними. У зв’язку з цим хромосомні хвороби являють собою нові мутації, що виникають у статевих клітинах батьків. Більшість ознак і хвороб людини залежать не від одного чи двох генів, а від цілого комплексу генів і в той же час від умов навколишнього середовища.
Мітохондрії мають власну ДНК (мтДНК); мтДНК розташовується в матриксі органели і представлена кільцевою хромосомою. Вважається, що при клітинному розподілі мітохондрії випадково розподіляються між дочірніми клітинами. Для мітохондріальних хвороб характерна різна експресивність (сила прояву), оскільки фенотипічний прояв патологічного гена залежить від співвідношення нормальних і мутантних мітохондрій. Серед мітохондріальних хвороб найкраще вивчений синдром Лебера. Мітохондріальні хвороби успадковуються тільки по материнській лінії.
Генні захворювання
Відомо приблизно чотири тисячі генних захворювань, характер успадкування яких визначається законами Менделя. Вони становлять численну та різноманітну за клінічною картиною групу патологій, основою яких є мутація одного гена.
Генні хвороби — спадкові патології, які спричинені мутацією одного гена і передаються наступним поколінням за законами Менделя.
Середня загальна частота новонароджених із такими хворобами становить 1% . Із них приблизно 50% уражені аутосомно-домінантними патологіями, 25% — аутосомно-рецесивними та 25% — зчепленими з Х-хромосомою. Захворювання, детерміновані генами, які містяться в У-хромосомі чи мітохондріях, трапляються дуже рідко. Хворобу вважають досить поширеною, якщо її частота сягає 1:10 000 новонароджених. За частоти ураження 1:11 000—40 000 новонароджених патологія має середню поширеність.
Аутосомно-домінантні патології
Найвідомішими аутосомно-домінантними захворюваннями є хорея Гентингтона, синдром Марфана, синдром Холта — Орама, нейрофіброматоз, серпоподібно-клітинна анемія, періодичний параліч. Характерна ознака цих патологій — порушення синтезу структурних або специфічних білків (наприклад, гемоглобіну).
Дія мутантного гена виявляється практично завжди. Хворі хлопчики та дівчатка народжуються з однаковою частотою.
Хорея Гентингтона також відноситься до генних захворювань.
Хорея Гентингтона обтяжена тим, що ознаки патології звичайно виявляються в середньому віці, коли багато хворих уже мають дітей. Після появи симптомів тривалість життя становить до 15 років. Це повільне згасання є додатковим джерелом переживань для хворих та їх рідних. Ген, що кодує хорею Гентингтона, домінантний, він завжди виявляється, тому якщо уражений один із батьків, вірогідність народження хворої дитини становить 50%.
Синдром Марфана.
Він полягає у системному ураженні сполучної тканини і характеризується високою пенетрантністю та різною експресивністю. Хворобу зумовлює мутація гена FBN1, локалізованого у довгому плечі хромосоми 15 (15^21.1). Виявлено велику кількість мутацій цього гена, що спричинює значну клінічну поліморфність захворювання. Ген FBN1 кодує синтез білка фібриліну, що є складовою сполучної тканини і забезпечує її пружність. Блокування синтезу цього білка призводить до підвищеного розтягання сполучної тканини.
Синдром Марфана вражає опорно-рухову, серцево-судинну системи та органи зору. Хворі мають характерний зовнішній вигляд: високий зріст, астенічну (кволу, слабку) статуру. Порушення опорно-рухової системи — це непропорційно довгі пальці (арахнодактилія — "павукові" пальці), видовжений череп, деформація грудної клітки (воронкоподібна або кілеподібна), викривлення хребта, надмірна рухомість суглобів, плоскостопість. Характерними порушеннями серцево-судинної системи є випинання мітрального клапана в бік лівого передсердя, розширення аорти у висхідному або черевному відділі з розвитком аневризми (випинання). Патологія органів зору полягає у короткозорості високого ступеня внаслідок підвивиху (або зсуву) кришталика та різному кольорі райдужної оболонки. Можуть траплятися також пахові, стегнові, діафрагмальні грижі, іноді — опущення нирок, емфізема легенів, ослаблення слуху аж до повної глухоти. Попри всі ці порушення, психічний і розумовий розвиток хворих відповідає нормі.
Тривалість життя хворого на синдром Марфана зумовлена ступенем ураження серцево-судинної системи і сягає в середньому 35 років.
Синдром Холта — Орама (синдром "рука—серце").
Він супроводжується множинними природженими вадами розвитку. Частоту захворювання поки що не визначено. Мутації гена ТВХ5, розташованого в довгому плечі хромосоми 12 (12^24,1), призводять до відсутності його продукту, внаслідок чого розвивається хвороба.
Клінічна картина синдрому Холта — Орама характеризується аномаліями верхніх кінцівок і природженими вадами серця. Дефекти розвитку рук варіюють від недорозвитку чи відсутності першого пальця кисті або його трьохфаланговості до недорозвитку або повної відсутності променевої кістки з формуванням променевої косорукості. Частіше вражається ліва рука. Спостерігаються й інші скелетні зміни: недорозвиток лопаток і ключиць, сколіоз (бокове викривлення хребта), воронкоподібна деформація грудини, викривлення мізинця, зрощення пальців, недорозвиток інших пальців кисті. У 50% хворих перший палець не протиставлений решті пальців кисті
У більшості хворих (до 85%) проявляються різні форми природжених вад серця: дефекти міжпередсердної та міжшлуночкової перегородок, відкрита артеріальна протока (за нормою наявна в кровоносній системі плоду), звуження аорти та легеневої артерії, випинання мітрального клапана в бік лівого передсердя тощо. Інтелект хворих на синдром Холта — Орама, як правило, зберігається. Прогноз життя залежить від тяжкості ураження серця.
Лікування синдрому Холта — Орама полягає у медикаментозному запобіганні розвитку інфекційних хвороб серця (наприклад, ендокардиту) та реконструктивній хірургії серцевих перегородок чи клапанів.

Також генними захворюваннями є муковісцидоз, фенілкетонурія, дальтонізм, гемофілія, серповидноклітинна анемія
Муковісцидоз – виникає внаслідок пошкодження гена, розташованого на хромосомі 7. Призводить до загущення секретів залоз зовнішньої секреції. Викликає значні проблеми в роботі травної і дихальної системи.
Фенілкетонурія – пошкодження гена, розташованого на хромосомі 12. Призводить до неможливості переробки амінокислоти фенілаланіну. За відсутності лікування призводить до ураження нервової системи.
Дальтонізм – пошкодження одного з трьох генів, що відповідають за синтез білка опсину, який сприймає відповідний колір. Ген синього опсину розташований на хромосомі 7, а гени червоного і синього на Х-хромосомі. Призводить до втрати можливості сприймати відповідний колір.
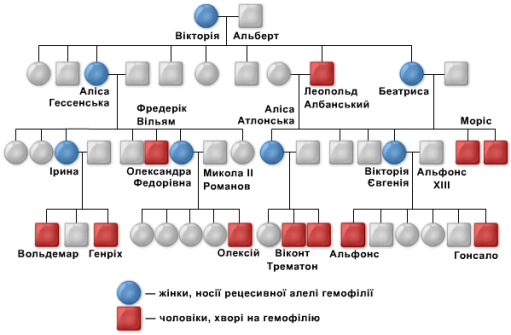
Гемофілія – пошкоджує один з генів, що відповідають за процес зсідання крові. Ці гени розташовані на Х-хромосомі. Спричинює порушення процесу зсідання крові. Гемофілія є прикладом зчепленого зі статтю генного захворювання. Вона стала добре відомою після того, як було встановлено успадкування цієї хвороби у нащадків англійської королеви Вікторії. Проте сама королева не була хворою, а була лише носієм.
Серповидноклітинна анемія – виникає в результаті заміни нуклеотиду А на Т в гені, який розташований на хромосомі 11. Внаслідок мутації є заміна в молекумі β-ланцюга гемоглобіну амінокислоти глутаміну на валін. Така молекула значно гірше транспортує кисень.
- Закріплення вивченого
Перегляд відео «Болезни и смерти королей»
https://www.youtube.com/watch?v=AHqVnjDdeqk
- Підсумок уроку
Завдяки прогресу медичної генетики і розширенню уявлень про характер успадкування різних захворювань та вплив факторів навколишнього середовища на появу мутантних генів стали набагато виразніші шляхи лікування і профілактики С.х. Радикальним методом лікування спадкових моногенних хвороб повинна стати генна терапія, однак лише в останні роки з’явилися реальні передумови для її практичного застосування.
- Домашнє завдання
Опрацювати текст параграфа
Завдання 6, 7 письмово


про публікацію авторської розробки
Додати розробку