Презентація Хромосомні хвороби людини


Хромосомні хвороби людини.

Генетика людини. У 1929 р. радянський генетик, невропатолог С. Н. Давіденко організував першу в світі медико-генетичну консультацію. Він першим в світі поставив питання про необхідність складання каталога генів людини, сформулював поняття про генетичну гетерогенність спадкових хвороб людини.

Будь які прояви життєдіяльності організму - результат взаємодії спадкових і середовищних факторів. Хвороба – це також результат взаємодії зовнішніх ушкоджуючих і внутрішних факторів.

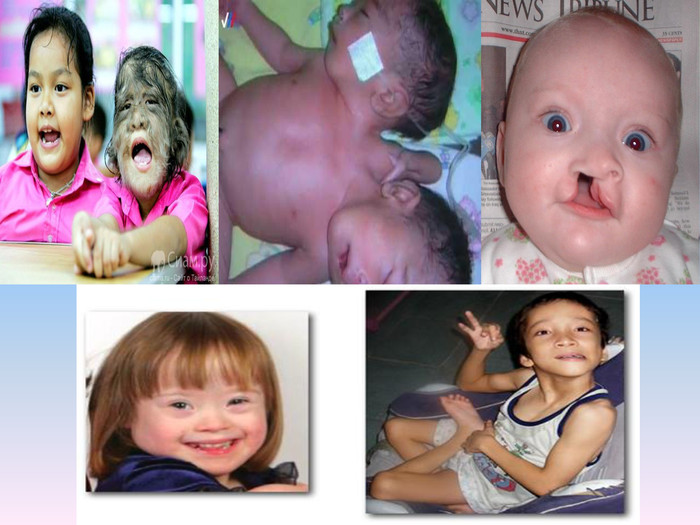
За даними медико – генетичної служби в Україні щорічно народжується біля 13 тис. новонароджених із урадженою патологією. З них 12.5тис із уродженими вадами розвитку і до 500 дітей із хромосомними хворобами, більшу частину,яких складає діти із Синдромом Дауна.
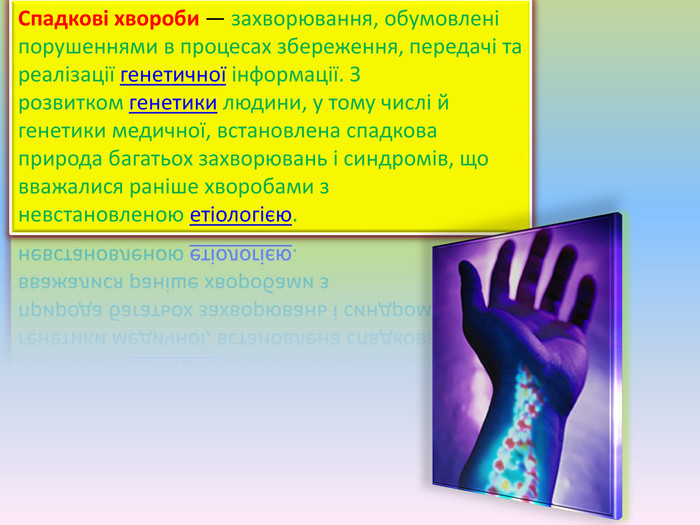
Спадкові хвороби — захворювання, обумовлені порушеннями в процесах збереження, передачі та реалізації генетичної інформації. З розвитком генетики людини, у тому числі й генетики медичної, встановлена спадкова природа багатьох захворювань і синдромів, що вважалися раніше хворобами з невстановленою етіологією.

Спадкові хвороби переслідували людство упродовж усієї його історії. Через непередбачуваність і «безпричинність» вони викликали у людей містичний страх. Їх сприймали як прокляття чи покарання богів.

Мутації – причини спадкових захворювань.
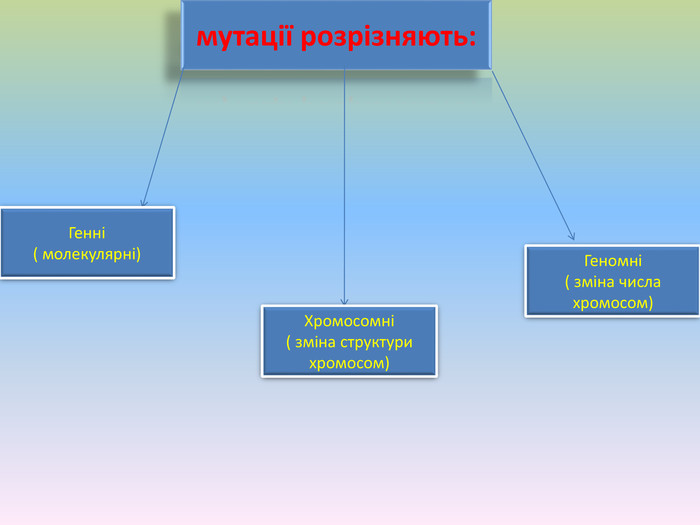
мутації розрізняють: Генні( молекулярні)Хромосомні( зміна структури хромосом)Геномні( зміна числа хромосом)

Хромосомні і генні хвороби. Хромосомні хвороби – це захворювання,пов'язані з геномними і хромосомними мутаціями. Як правило при хромосомних хворобах порушується: Збалансованість набору генів. Сувора детермінованість нормального розвитку організму. Це призводить до внутрішньочеревної загибелі ембріонів і плодів.

Генні хвороби викликаються генними мутаціями ( молеку-лярними змінами на рівні ДНК)Більшість спадкових захворювань – це генні хвороби.

Останньою яскравою перемогою генетиків стало народження на початку січня цього року у Великій Британії дівчинки без спадкової аномалії, через яку три покоління жінок по лінії її батька хворіли на рак грудей. До появи хвороби призводив мутований ген BRCA, який у нормі придушує ріст ракових клітин. Ймовірність його успадкування від батька становила 50%. Через такий збій у генах ризик розвитку раку грудей у жінок збільшується на 80%, а раку яєчників – на 60%.

Проте медики змогли перервати цю сумну історію роду. Дівчинка з’явилася на світ шляхом штучного запліднення. У клініці Університетського коледжу Лондона створили 11 ембріонів. На третій день розвитку, коли кожен з них складався з восьми клітин, було проведено генетичний аналіз ембріонів на наявність мутованого гена BRCA. Для цього лікарі взяли з кожного ембріона по одній клітині, що не завдає їм шкоди, оскільки клітини ще недиференційовані. В організм матері було пересаджено два ембріони з нормальним геном BRCA, один з яких прижився.

Хромосомні хвороби – це велика група уроджених спадкових захворювань, що клінічно характеризуються множинними уродженими вадами розвитку.

Точне число хромосом у клітинах людини було визначено в 1956році ( Tjio Levan)

Хвороба, що найчастіше зустрічається,трисомія 21,клінічна була описана у 1866р. англіським педіатром Джоном Ленгдоном Дауновим ( за його ім’ям і названо цю хворобу – синдром Дауна)

Перший клінічний опис синдрома моносомії по Х – хромосомі ( 45, ХО), як окремої форми хвороби було зроблено російським клініцистом М. О. Шерешевський у 1925р. Та Генрі Тернером.

Аномалії в системі статевих хромосом у чоловіків (трисомія XXY) як клінічний синдром упереше описав Гаррі Клайнфельтер у 1942р.

Ці три захворювання стали об’єктом перших клініко - цитогенетичних досліджень проведенних у 1959р. Розшифровка етіології синдромів Дауна, Шерешевського –Тернера і Клайнфельтера. Відкрили новий розділ у медицині – хромосомні хвороби.
Хромосомні хвороби виникають внаслідок мутації у статевих клітинах одного з батьків.

Геномні мутаціїГеномні мутації призводять до змін кількості хромосом у наборі:поліплоїдія – збільшення кількості хромосом, кратного гаплоїдному набору (46+23+23…+23); аутоплоїдія – виникає як результат поділу хромосом без подальшого поділу клітини; анеуплоїдія – зміна кількості хромосом в диплоїдному наборі, некратному гаплоїдному (2n+1; 2n-1)46+1,+2……-1,-2……….

Хромосомні мутації Хромосомні мутації Аберації – виникаютьв результаті значних змін в структурі хромосом. До них належать: нехватки; делеції; дуплікації; інверсії; транслокації; транспозиції.
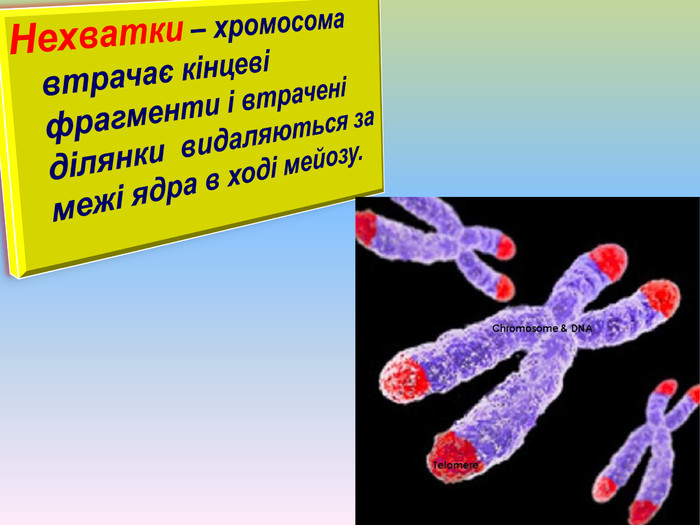
Нехватки – хромосома втрачає кінцеві фрагменти і втрачені ділянки видаляються за межі ядра в ході мейозу.

Делеції – втрата частини хромосоми всередині. Це викликає зміну фенотипу, але якщо втрачені гени є життєво необхідними, то це призводить до смерті або патології.
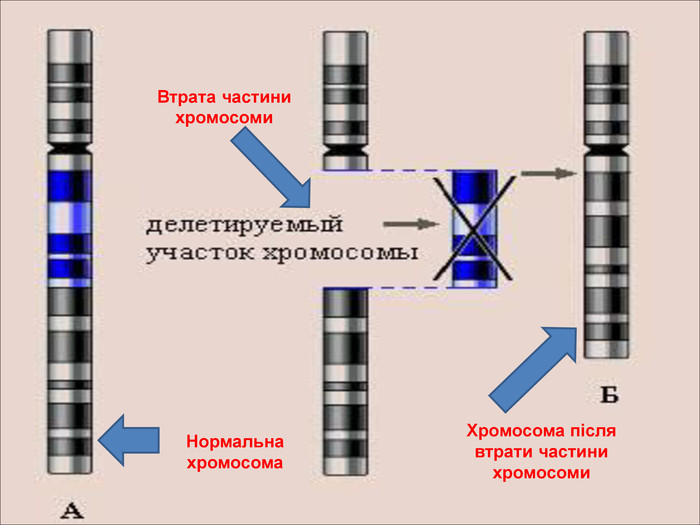
Втрата частини хромосоми Нормальна хромосома. Хромосома після втрати частини хромосоми

Дуплікації – подвоєння якої завгодно ділянки хромосоми, що впливає в основному на фенотип, бо гени лише додаються, а не втра-чаються. Мутації виникають тому, що змінюється РНК.
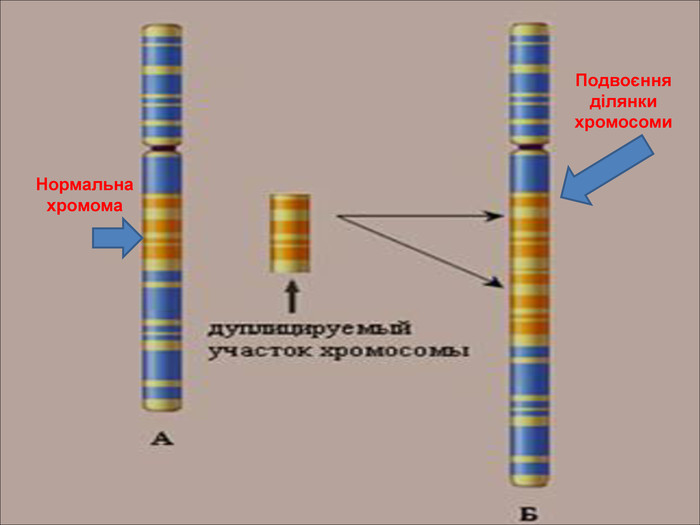
Нормальна хромома. Подвоєння ділянки хромосоми

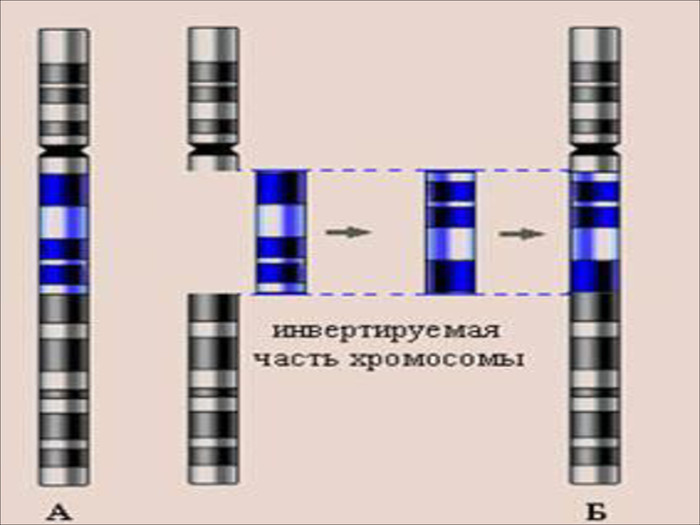
Інверсії – зміна порядку розміщення генів в хромосомі. Вони виникають в результаті двох розривів хромосоми, при цьому фрагмент повертається на місце, але повернувшись на 180 градусів кількість генів не змінюється, тому особливого впливу на організм такі мутації не здійснюють.

Транслокації – мутації, що пов’язані з: -обміном ділянками між негомологічними хромосомами; -прикріплення ділянки однієї хромосоми до хромосоми негомологічної пари. Транспозиції – це: вставка невеликого фрагменту хромосоми, що несе декілька генів в якусь іншу ділянку хромосоми;перенесення частини генів в інше місце.
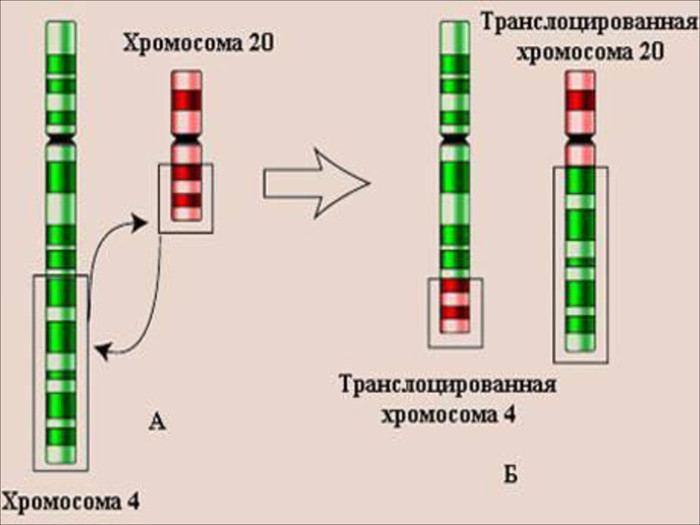

Генні мутації Генні (крапкові) мутації – стійкі зміни окремих генів, що виникають в результаті: - заміни однієї або декількох азотистих основ уструктурі ДНК на інші;- випадання деяких азотистих основ; - доповнення нових азотистих основ. Це призводить до порушення порядку списування інформації. Генні мутації змінюють морфологічні, біохімічні і фізіологічні властивості організму.

Генні мутаціїГенні мутації не проявляються на зовнішнійморфології хромосом, їх не можна побачити підмікроскопом. Коли змінюється порядок нуклеотидів в ДНК хромосоми, то відбуваються зміни і молекули РНК, яка будується на основі ДНК. Якщо змінюється РНК, то змінюється і білок, який будується на основі РНК. Отже, виникає мутація.
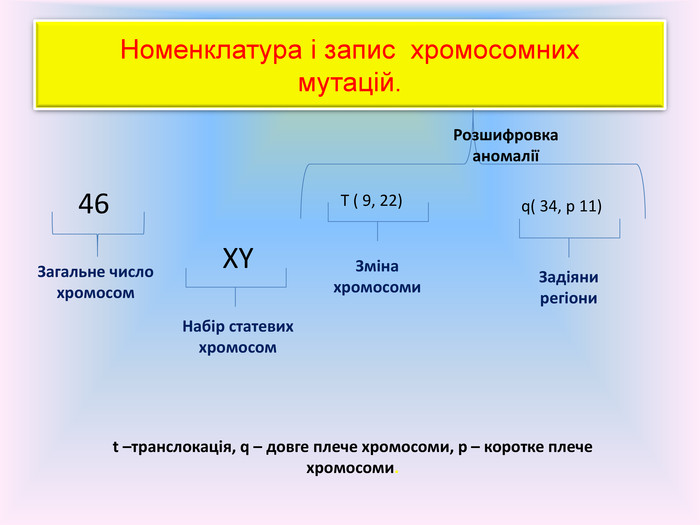
Номенклатура і запис хромосомнихмутацій.46 Загальне число хромосом. XYНабір статевих хромосом. T ( 9, 22)q( 34, р 11)Зміна хромосоми. Задіяни регіони. Розшифровка аномаліїt –транслокація, q – довге плече хромосоми, р – коротке плече хромосоми.
Хоча геномні мутації в тваринному та рослинному світі різноманітні. У людини виявлено лише 3 типи геномних мутацій: Тетраплоїдія. Триплоїдія анеуплоїдія

Анеуплоїдія - стан клітини, тканини чи організму, при якому одна чи декілька хромосом звичайного набору або відсутня, або представлена додатковими копіями. Уперше анеплоїдію у людини виявили у 1959р. Дж. Леже і Р. Турпін. Приклади анеуплоїдії у людини: синдром Дауна — трисоміясиндром Кляйнфельтера синдром Тернера

Множинність ураження – загальне для усіх форм хромосомних хвороб. Це - 1. Черепно - лицьові дизморфії.2. Уроджені вади розвитку внутрішніх органів і частин тіла.3. Відставання у психічному розвитку.4. Порушення функцій нервової, ендокринної і імунної системи.

Синдром Дауна (трисомія 21 хромосоми)Це одна з найпоширеніших патологій людини. З частотою на світі дітей з цим недугом складає один на 800-900 новонароджених. Синдром Дауна (трісомія по хромосомі 21) - одна з форм патології генома, при якій всього каріотип представлений 47 хромосомами замість нормальних 46, оскільки хромосоми 21-ої пари, замість нормальних двох, представлені трьома копіями. Існує ще дві форми даного синдрому: транслокація хромосоми 21 на інші хромосоми (частіше на 15, рідше на 14, ще рідше на 21, 22 і Y-хромосому) - 4 % випадків , і мозаїчний варіант синдрому - 5%.

Синдром отримав назву на честь англійського лікаря Джона Дауна (John Down), що вперше описав його в 1866 році. Зв'язок між походженням природженого синдрому і зміною кількості хромосом був виявлений лише в 1959 році французьким генетиком Жеромом Леженом.
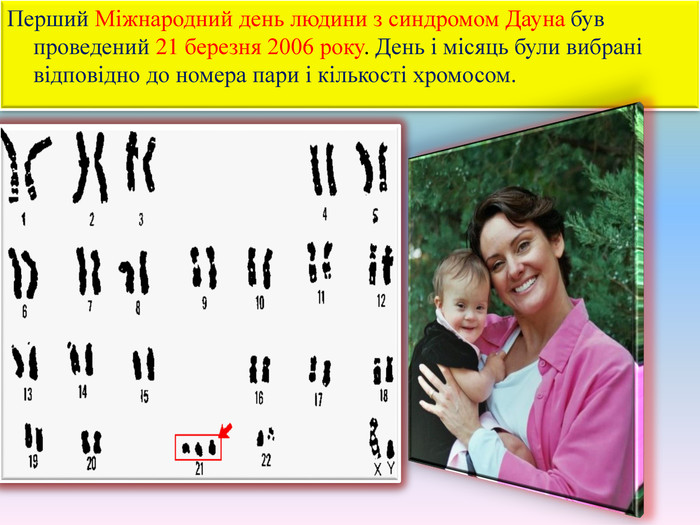
Перший Міжнародний день людини з синдромом Дауна був проведений 21 березня 2006 року. День і місяць були вибрані відповідно до номера пари і кількості хромосом.

Історія. Англійський лікар Джон Ленгдон Даун перший пише і характеризує синдром Дауна як форму психічного розладу в 1862 році і стає відомішим, опублікувавши свою доповідь в 1866 році. Із-за епікантуса Даун використовував термін монголоїди (синдром же називали «монголізмом»). Уявлення про синдром Дауна було дуже прив'язане до расизму аж до 1970-х років. До середини XX століття причини синдрому Дауна залишалися для всіх невідомими, проте був відомий взаємозв'язок між вірогідністю народження дитяти з синдромом Дауна і віком матері, також було відомо те, що синдрому були схильні всі раси. Існувала теорія про те, що синдром викликаний поєднанням генетичних і спадкових чинників. Інші теорії дотримувалися думки, що він викликаний травмами під час пологів.

З відкриттям у 1950-х роках технологій , що дозволяють вивчати каріотип, стало можливо визначити аномалії хромосом, їх кількість і форму. У 1959 році Жером Лежен виявив, що синдром Дауна виникає через трисомії 21-ї хромосоми. У 1961 році вісімнадцять генетиків написали редактора «The Lancet», що Монгольський ідіотизм «вводить конотації в оману» і що це «незграбний термін» і він повинен бути змінений. «The Lancet» підтримує назву «синдром Дауна». Всесвітня організація охорони здоров'я (ВООЗ) офіційно прибрала назву «монголізм» у 1965 році після звернення монгольських делегатів. Однак навіть 40 років потому, назва «монголізм» з'являється у провідних медичних посібниках, наприклад, у «повсюдних і систематичних патологіях» 4-го видання (2004), під редакцією професора сера Джеймса Андервуда. Захисники прав хворих та батьки хворих вітали ліквідацію монголоїдного ярлика, повішеного на їхніх дітей. Перша група у США, монголоїдний Рада Розвитку, змінила свою назву на Національна асоціація «синдрому Дауна» в 1972 році.
Епідеміологія. Синдром Дауна не є рідкісною патологією - в середньому спостерігається один випадок на 700 пологів; в даний момент, завдяки пренатальній діагностиці, частота народження дітей з синдромом Дауна зменшилася до 1 до 1100. У хлопчиків і у дівчаток аномалія зустрічається з однаковою частотою 1:1. Частота народжень дітей з синдромом Дауна 1 на 800 або 1000. У 2006 році Центр з контролю і профілактики захворювань оцінив як один на 733 живонароджених у США (5429 нових випадків на рік). Близько 95% з них по трисомії 21-ї хромосоми. Синдром Дауна зустрічається в усіх етнічних групах і серед всіх економічних класів.
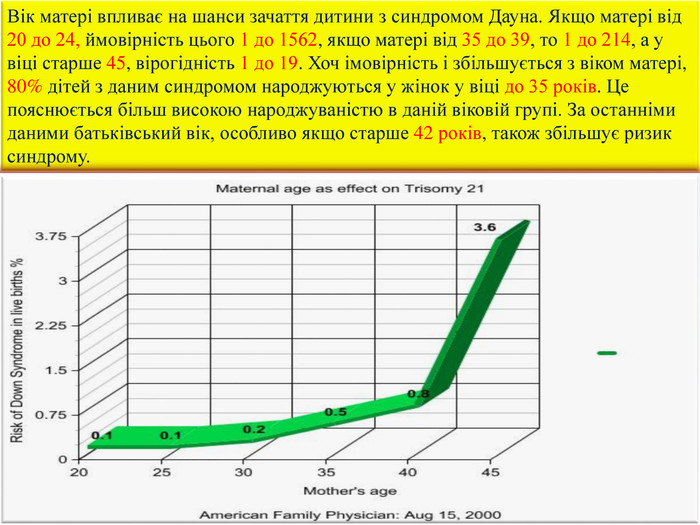
Вік матері впливає на шанси зачаття дитини з синдромом Дауна. Якщо матері від 20 до 24, ймовірність цього 1 до 1562, якщо матері від 35 до 39, то 1 до 214, а у віці старше 45, вірогідність 1 до 19. Хоч імовірність і збільшується з віком матері, 80% дітей з даним синдромом народжуються у жінок у віці до 35 років. Це пояснюється більш високою народжуваністю в даній віковій групі. За останніми даними батьківський вік, особливо якщо старше 42 років, також збільшує ризик синдрому.

Цитогенетичні варіанти Синдрома Дауна різноманітні. Трісомія - це наявність трьох гомологічних хромосом замість пари в нормі. Синдром Дауна і подібні хромосомні аномалії частіше зустрічаються у дітей, народжених немолодими жінками. Точна причина цього невідома, але, мабуть, вона якось пов'язана з віком яйцеклітин матері. Трісомія відбувається через те, що під час мейозу хромосоми не розходяться. При злитті з гаметою протилежної статі у ембріона утворюється 47 хромосом, а не 46, як без трисомії. Трісомія 21-ї хромосоми у 95% випадків є причиною виникнення синдрому Дауна, і у 88% випадків через нерозходження материнських гамет та у 8% - чоловічих.

Каріотип для Трисомії при синдромі Дауна.

Мозаїцизм Трісомія зазвичай викликана нерозходженням хромосом при формуванні статевих клітин батьків (гамет), в цьому випадку всі клітини організму дитини будуть нести аномалію. При мозаїцизмі ж ннерозходження виникає в клітці зародка на ранніх стадіях його розвитку, в результаті чого порушення каріотипу зачіпає тільки деякі тканини і органи. Даний варіант розвитку синдрому Дауна називається «мозаїчний синдром Дауна» (46, XX/47, XX, 21). Дана форма синдрому є як правило більш легкої (в залежності від просторості змінених тканин та їх розташування в організмі), однак більше важка для пренатальної діагностики. По даному типу синдром з'являється в 1-2% випадків.

Робертсонівські транслокації Додатковий матеріал 21-ої хромосоми, що викликає синдром Дауна, може з'явитися за рахунок робертсонівських транслокацій в каріотипі одного з батьків. У даному випадку довге плече 21-ї хромосоми прикріплено до плеча іншої хромосоми (частіше за все 14-й [45, XX, дер (14; 21) (q10; q10)]). Фенотип у людини з робертсонівськіми транслокаціями відповідає нормі. Під час репродукції, нормальний мейоз підвищує шанс на трисомії 21-ї хромосоми і народження дитини з синдромом Дауна. Транслокації з синдромом Дауна часто називають (сімейний синдром Дауна). Це не залежить від віку матері та показує скоріше рівну роль батьківських організмів в появі синдрому Дауна. Даний тип появи синдрому займає 2-3% від усіх випадків.
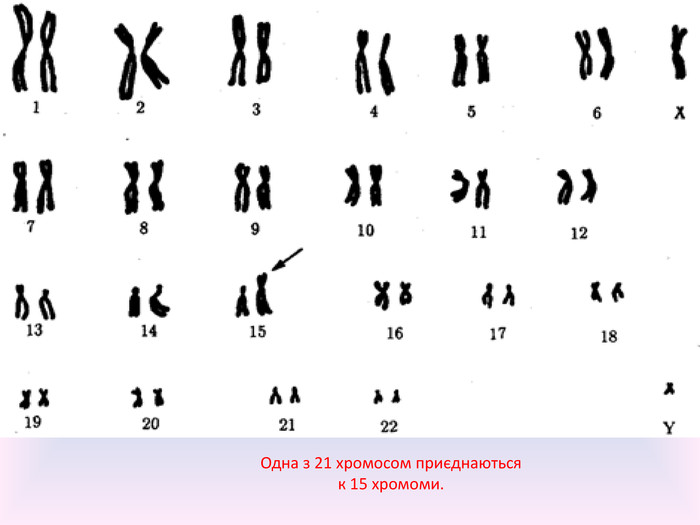
Одна з 21 хромосом приєднаються к 15 хромоми.

Хворі зазвичай невисокого зросту, відрізняються слабоумством. У них характерна зовнішність: монголоїдний розріз очей кругле сплощене обличчя короткий ніспласке переніссяепікант (напівмісяцева вертикальна складка у внутрішнього кута ока) маленькі деформовані вуханапіввідкритий рот із ледь висунутим язиком і нижньою щелепою, що висувається
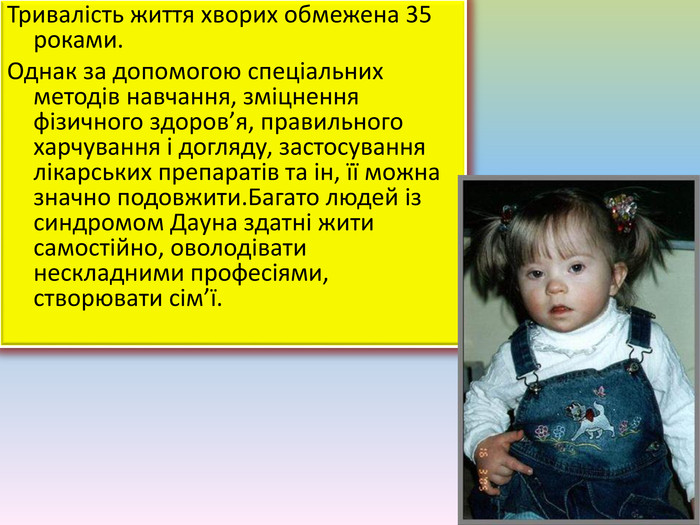
Тривалість життя хворих обмежена 35 роками. Однак за допомогою спеціальних методів навчання, зміцнення фізичного здоров’я, правильного харчування і догляду, застосування лікарських препаратів та ін, її можна значно подовжити. Багато людей із синдромом Дауна здатні жити самостійно, оволодівати нескладними професіями, створювати сім’ї.

Синдром Едвардса (трисомія з 18 хромосомою)Описано в 1960 році Джоном Едвардсом (John H. Edwards). Популяційна частота приблизно 1:7000. Для жінок старше 45 років ризик народити хвору дитину становить 0,7%. Дівчатка із синдромом Едвардса народжуються в три рази частіше за хлопчиків.
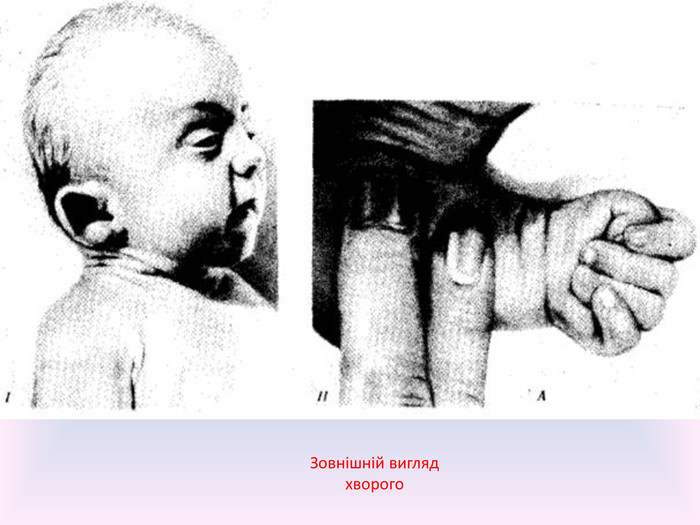
Зовнішній вигляд хворого
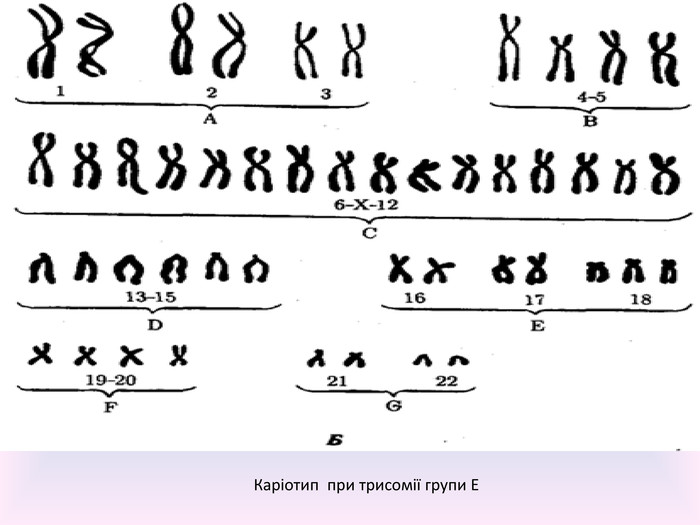
Каріотип при трисомії групи Е

Причини захворювання: Причиною захворювання : є наявність додаткової 18-ої хромосоми (трьох замість двох у нормі для диплоїдного набору) в каріотипі зиготи. Зайва хромосома зазвичай з'являється до запліднення. У людини нормальні статеві клітини - гамети - містять по 23 хромосоми (гаплоїдний набір) і, зливаючись, вони дають каріотип зиготи - 46 хромосом. До появи зайвої хромосоми у гамет зазвичай призводить неросходження хромосом при мейотичному розподілі, внаслідок чого статевій клітині надається 24 хромосоми. У разі, якщо така клітина зустріне при заплідненні гамету від протилежної статі, вони утворюють зиготу з трисомією. В одному випадку з десяти спостерігається мозаїцизм в явищі трисомії 18: зайву хромосому несуть не всі клітини організму. Це говорить про те, що неросходження сталося на ранній стадії розвитку зародка, а всі клітини з трисомією - нащадки клітини зародка яка неправильно поділилась.

Прояви синдрому. Діти у яких трисомія 18 народжуються з низькою , в середньому 2177 г, вагою. Найчастіше виникають аномалії мозкового та лицевого черепа. Нижня щелепа і ротовий отвір маленькі. Очні щілини вузькі і короткі. Вушні раковини деформовані і в переважній більшості випадків розташовані низько, кілька витягнуті в горизонтальній площині. Зовнішній слуховий прохід звужений, іноді відсутній. Грудина коротка, через що міжреберні проміжки зменшені і грудна клітина ширша і коротша нормальної.
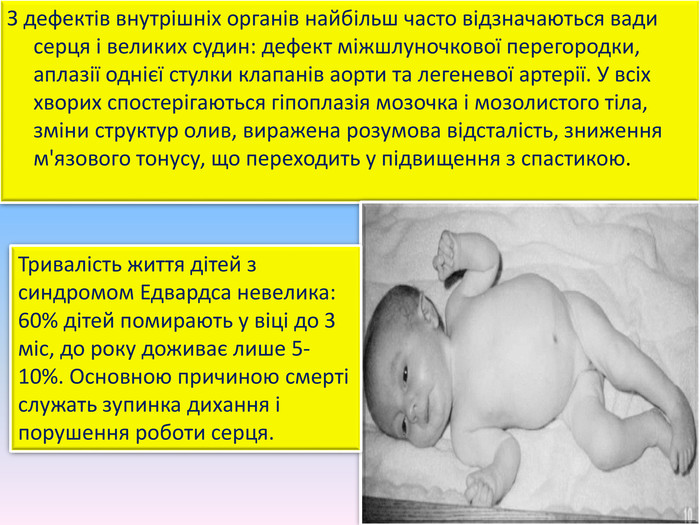
З дефектів внутрішніх органів найбільш часто відзначаються вади серця і великих судин: дефект міжшлуночкової перегородки, аплазії однієї стулки клапанів аорти та легеневої артерії. У всіх хворих спостерігаються гіпоплазія мозочка і мозолистого тіла, зміни структур олив, виражена розумова відсталість, зниження м'язового тонусу, що переходить у підвищення з спастикою. Тривалість життя дітей з синдромом Едвардса невелика: 60% дітей помирають у віці до 3 міс, до року доживає лише 5-10%. Основною причиною смерті служать зупинка дихання і порушення роботи серця.
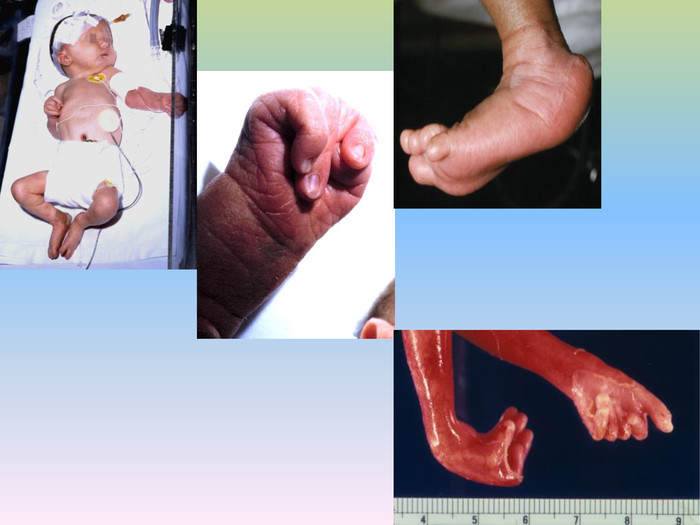
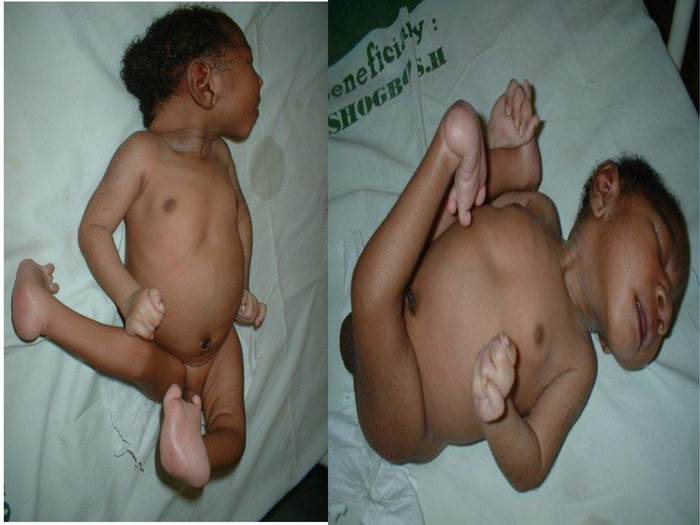
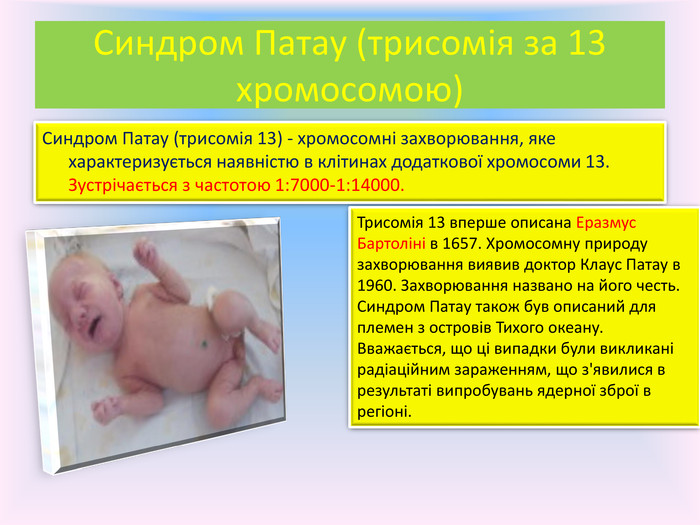
Синдром Патау (трисомія за 13 хромосомою)Синдром Патау (трисомія 13) - хромосомні захворювання, яке характеризується наявністю в клітинах додаткової хромосоми 13. Зустрічається з частотою 1:7000-1:14000. Трисомія 13 вперше описана Еразмус Бартоліні в 1657. Хромосомну природу захворювання виявив доктор Клаус Патау в 1960. Захворювання названо на його честь. Синдром Патау також був описаний для племен з островів Тихого океану. Вважається, що ці випадки були викликані радіаційним зараженням, що з'явилися в результаті випробувань ядерної зброї в регіоні.

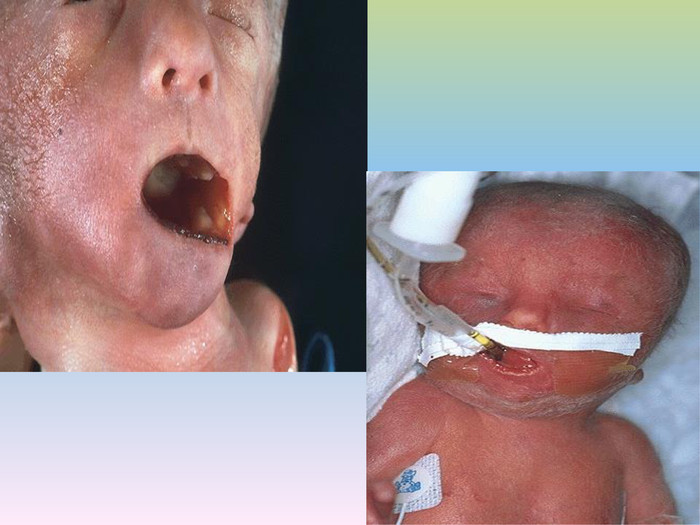
При синдромі Патау спостерігаються важкі вроджені вади. Діти з синдромом Патау народжуються з масою тіла нижче норми . У них виявляються, порушення розвитку різних відділів ЦНС, низький скошений лоб, звужені очні щілини, запали перенісся, деформовані вушні раковини, ущелина верхньої губи і піднебіння, полідактилія , коротка шия. У 80% новонароджених зустрічаються пороки розвитку серця: дефекти міжшлуночкової та міжпередсердної перегородок, транспозиції судин і ін. Спостерігаються фіброкістозні зміни підшлункової залози, додаткові селезінки, ембріональна пупкова грижа. Нирки збільшені, мають підвищену дольчатість , виявляються вади розвитку статевих органів. Для СП характерна затримка розумового розвитку.


У 80-85% хворих виявляється глухота. У зв’зку із тяжкими вадами розвитку 95% хворих дітей помирають у перші тижні або місяці (95% —до 1 року). Однак деякі хворі живуть декілька років.

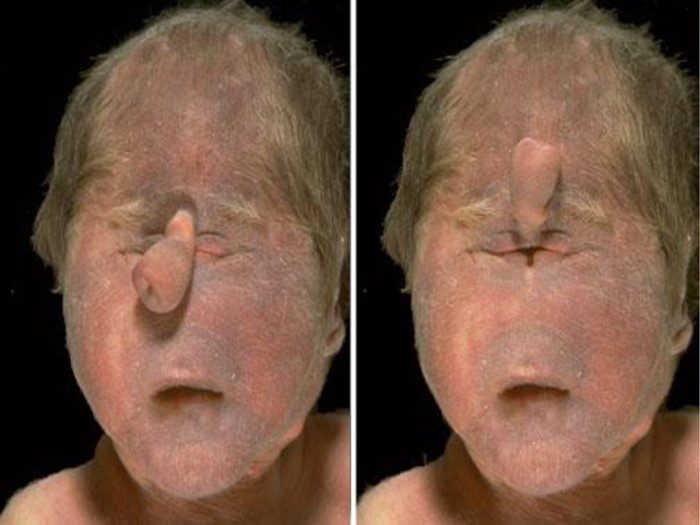

Синдром кошачого крику: Синдром кошачого крику (синоніми: хвороба котячого крику, синдром Лежена на ім'я описав його в 1963 р. французького вченого).
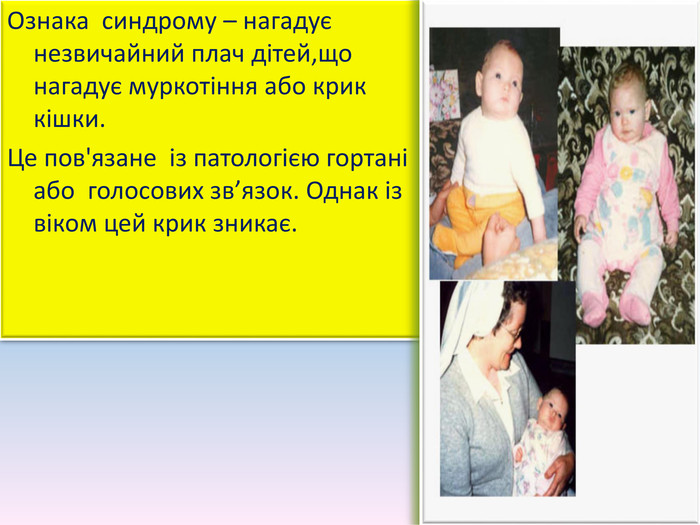
Ознака синдрому – нагадує незвичайний плач дітей,що нагадує муркотіння або крик кішки. Це пов'язане із патологією гортані або голосових зв’язок. Однак із віком цей крик зникає.

Характерний зовнішній вигляд хворих: - місяцеподібне обличчя (лялькове)- мікрогенія (малі розміри верхньої щелепи)- епікант- високе піднебіння- плоска спинка носу-косоокість (страбізм)- вушні раковини розташовані низько і деформовані.

Відзначають також вроджені вади серця, патологію кістково - м’язової системи, синдактилію стоп (повне аб часткове зрощення сусідніх пальців),плоскостопість, клешоногість та ін.), м’язову гіпотонію. Більшість дітей помирає у ранньому віці. Разом із тим відомі випадки життя хворих понад 50 років. Популяційна частота синдрому "кошачого крику" 1:40 000 - 1:50 000новонароджених
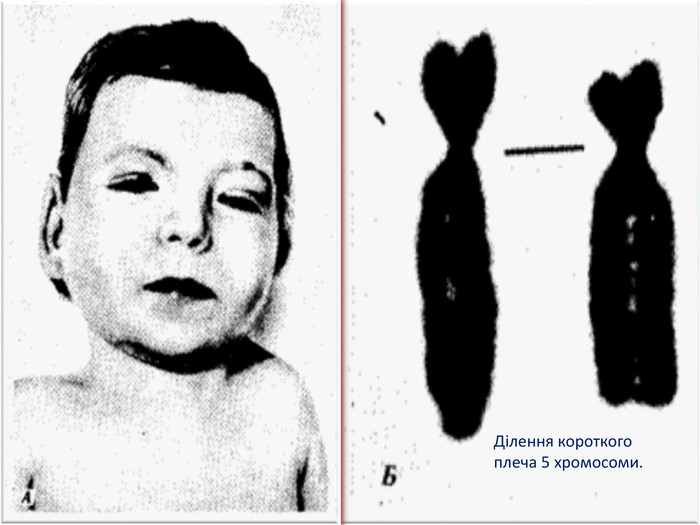
Ділення короткого плеча 5 хромосоми.

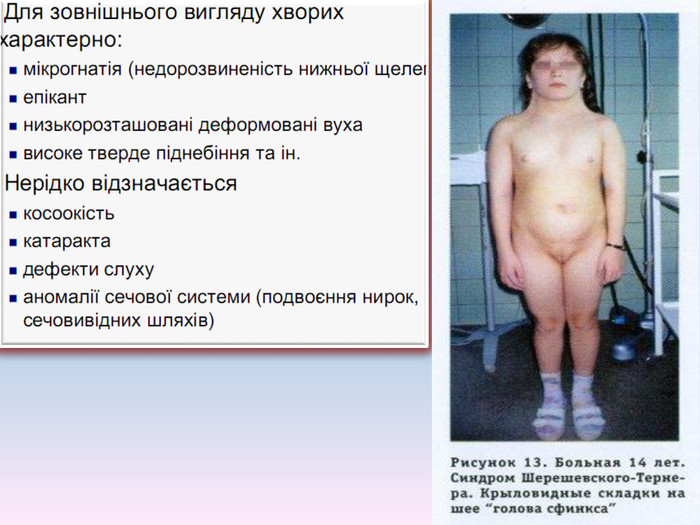
Синдром Шерешевського-Тернера Уперше описаний М. Шерешевським у 1925 р. і Тернером у 1938 р. Причина хвороби - порушення розходження статевих хромосом. Хворіють лише жінки, у них відсутня одна Х-хромосома (45, Х0) - єдина форма моносомії у живонароджених. Частота синдрому 1 : 3000 новонароджених дівчаток. Лише у 20% жінок вагітність хворим плодом зберігається до кінця і народжується жива дитина. В інших випадках відбувається спонтанний аборт абомертвонародження .
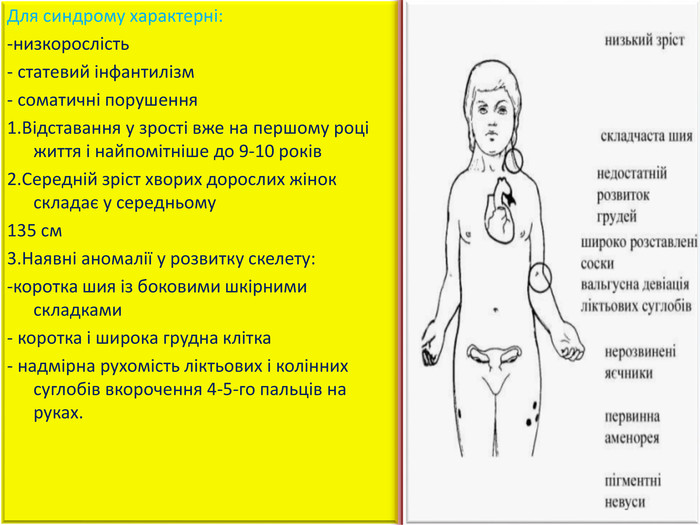
Для синдрому характерні: -низкорослість- статевий інфантилізм- соматичні порушення1. Відставання у зрості вже на першому році життя і найпомітніше до 9-10 років2. Середній зріст хворих дорослих жінок складає у середньому 135 см3. Наявні аномалії у розвитку скелету: -коротка шия із боковими шкірними складками- коротка і широка грудна клітка- надмірна рухомість ліктьових і колінних суглобів вкорочення 4-5-го пальців на руках.

Дівчина до і після операції.
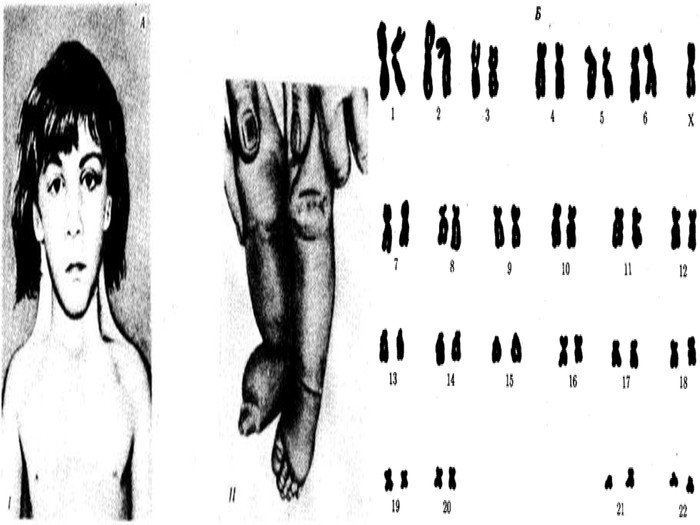


Тільця Барра в діагностиці хромосомних аномалій. До цитогенетичних методів, що викорис-товуються в клініці, належать: - визначення статевого хроматину (Х- і У-хроматину) в інтерфазних ядрах різних тканин- морфологічних особливостей хроматину в нейтрофілах периферичної крові (барабанні палички)- дослідження хромосом на стадії метафази мітозу для визначення каріотипу
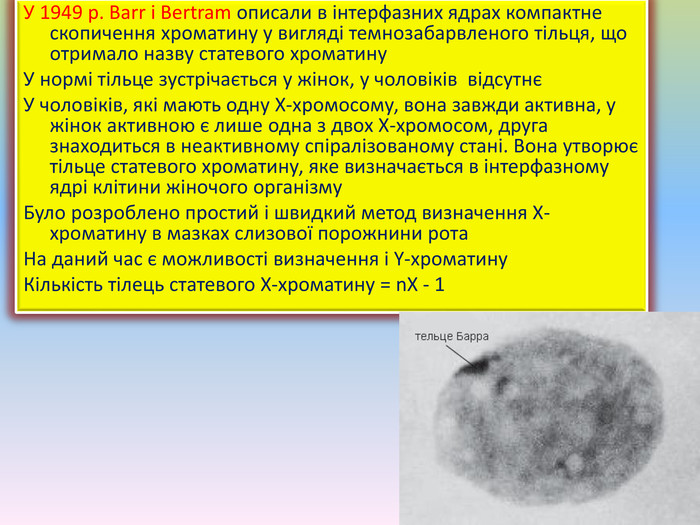
У 1949 р. Barr і Bertram описали в інтерфазних ядрах компактне скопичення хроматину у вигляді темнозабарвленого тільця, що отримало назву статевого хроматину. У нормі тільце зустрічається у жінок, у чоловіків відсутнєУ чоловіків, які мають одну Х-хромосому, вона завжди активна, у жінок активною є лише одна з двох Х-хромосом, друга знаходиться в неактивному спіралізованому стані. Вона утворює тільце статевого хроматину, яке визначається в інтерфазному ядрі клітини жіночого організму. Було розроблено простий і швидкий метод визначення Х-хроматину в мазках слизової порожнини рота. На даний час є можливості визначення і Y-хроматину. Кількість тілець статевого Х-хроматину = n. Х - 1

Полісомії за статевимихромосомами. Велика група хромосомних хвороб, представлена різними комбінаціям додаткових Х- або Y-хромосом. Загальна частота полісомій по Х- або Y-хромосомах серед новонароджених становить 1,5:1000—2:1000 В основному це полісомії XXX, XXY и XYYМозаїчні форми складають приблизно 25%

Синдром полісомії за Х-хромосомою у жінок(синдром «суперсамки»)Синдром включає- трисомію (каріотип 47, XXX)- тетрасомію (48, ХХХХ)- пентасомію (49, ХХХХХ)Найчастіше зустрічається трисомія — 1 на 1000 дівчаток, що народилися. Відзначається незначне зниження інтелекту, підвищена імовірність розвитку психозів і шизофренії із несприят-ливим перебігом. Можлива девіантна статева поведінка. Здатність народжувати дітей у таких жінок страждає у меншій мірі .

Із збільшенням числа додаткових Х-хромосом-зростає ступень відхилення від норми-У жінок із тетра- і пентасомією описані відхилення у розумовій розвиненості- черепно-лицьові дизморфії-аномалії зубів, скелета і статевих органів. Жінки навіть із тетрасомією за Х-хромосомою можуть мати дітей. Діагностика синдрому полісомії X включає визначення статевого хроматину і дослідження каріотипу хвороїРаціонального лікування немає.

Синдром Клайнфельтера. Описаний Клайнфельтером у 1942 р. Хворіють лише хлопчики. Частота — 2 із 1000 новонароджених хлопчиків. Хворі мають зайву Х-хромосому (каріотип 47, XXY)Поряд із цим, зустрічаються варіанти полісомії із великою кількістю Х- і Y-хромосом, які також відносять до синдрому Клайнфельтера До народження захворювання практично не діагностується. Генетичні аномалії проявляються в період статевого дозрі-вання у вигляді недорозвиненості сім’яників і вторинних статевих ознак.
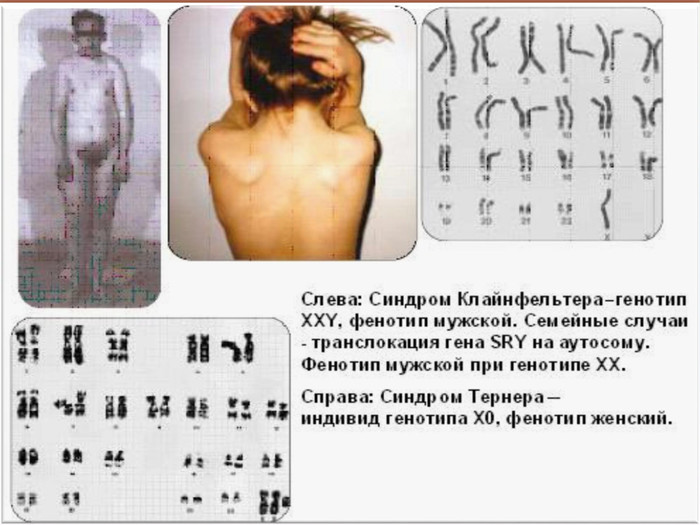
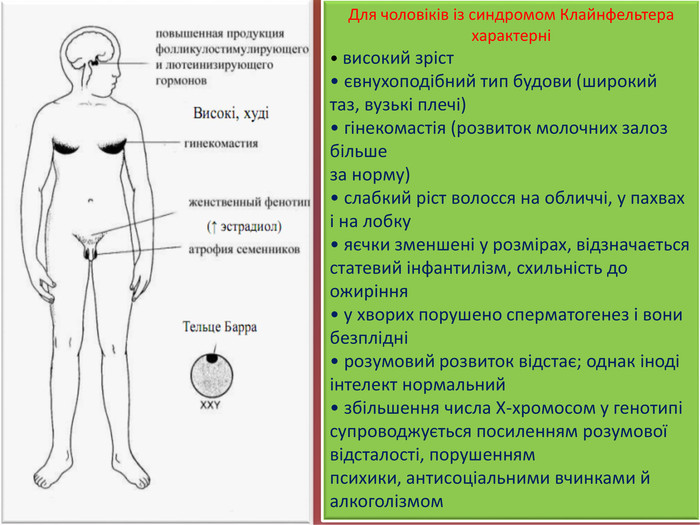
Для чоловіків із синдромом Клайнфельтера характерні• високий зріст• євнухоподібний тип будови (широкий таз, вузькі плечі)• гінекомастія (розвиток молочних залоз більшеза норму)• слабкий ріст волосся на обличчі, у пахвах і на лобку• яєчки зменшені у розмірах, відзначається статевий інфантилізм, схильність до ожиріння• у хворих порушено сперматогенез і вони безплідні• розумовий розвиток відстає; однак іноді інтелект нормальний• збільшення числа Х-хромосом у генотипі супроводжується посиленням розумової відсталості, порушенням психики, антисоціальними вчинками й алкоголізмом

Синдром дисомії за. Y-хромосомою (47, XYY)Описаний у 1961 р. Зустрічається з частотою 1 на 1000 новонароджених хлопчиків. Чоловіки із набором хромосом 47, XYY не відрізняються від норми за фізичним і розумовим розвитком. Відзначається невелике збільшення зросту — біля 185 смІноді спостерігається незначне зниження інтелекту, схильність до агресивних і антисоціальних вчинків. За деякими даними, у місцях позбавлення волі чоловіків із генотипом XYY у 10 разів більше, ніж чоловіків із нормальним генотипом

Дякуємо за увагу!


про публікацію авторської розробки
Додати розробку
-

Атмажова Наталя Сергіївна
21.10.2021 в 13:26
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
-

Раєва Алла Фахраддинівна
13.03.2020 в 17:00
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
-

Лєднєва Наталія Юріївна
23.02.2020 в 14:49
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
-

Товстая Тетяна
27.09.2019 в 21:18
Загальна:
5.0
Структурованість
5.0
Оригінальність викладу
5.0
Відповідність темі
5.0
Показати ще 1 відгук